吞噬功能缺陷病
吞噬功能缺陷病百科
吞噬功能缺陷病,包括慢性肉芽腫病和Chediak-Higashi綜合征,吞噬功能缺陷將導致機體對病原微生物的易感性增高,常發生各種化膿菌感染及條件致病菌的感染.原發性吞噬功能缺陷系指先天性或遺傳性的內源性吞噬功能不全.
吞噬功能缺陷病
吞噬功能缺陷病病因
(一)發病原因
吞噬細胞的功能不足的病因,多為遺傳性疾病,如CGD主要是伴性型染色體遺傳性疾病,但也可以常染色體隱性方式遺傳,為此,女孩也可發病,因此患兒白細胞在實驗室試驗時,可證明吞噬細菌的白細胞耗氧量不增加,不形成過氧化物或過氧化氫,細菌不被殺死,同時還存在白細胞缺乏過氧化物酶,或缺乏6-磷酸葡萄糖脫氫酶,或缺乏丙酮酸鹽激酶等,缺乏6-磷酸葡萄糖脫氫酶,不能殺傷過氧化氫酶陽性的細菌;缺乏丙酮酸鹽激酶,則喪失殺傷葡萄球菌的能力,慢性肉芽腫病為X連鎖隱性遺傳,少數為常染色體隱性遺傳,Chédiak-Higashi綜合征為常染色體隱性基因的遺傳病,其病因還不很清楚.
(二)發病機制
慢性肉芽腫病為X連鎖隱性遺傳,少數為常染色體隱性遺傳,Chédiak-Higashi綜合征為常染色體隱性基因的遺傳病,其發病機制還不明.
吞噬細胞缺陷發病機制可能有,中性粒細胞減少癥,吞噬細胞的功能不足.
1.中性粒細胞減少癥
先天性遺傳性中性粒細胞減少癥罕見,這類患兒多形核白細胞數低於5.0×108/L,表現瞭頻繁感染,周期性中性粒細胞減少癥,則按15~35天為一周期(平均21天)的反復減少,減少期為數天,數量可下降至5.0×107/L,伴有胰腺不全的中性粒細胞減少癥(Shwachman綜合征或全身播散性肉樣瘤),其患兒發育不良,呈現脂肪痢及反復感染等癥狀,中性粒細胞數降低到2.0~4.0×108/L,患者常伴有輕度糖尿病,這類患兒對病毒感染預後不良,伴有低免疫球蛋白的中性粒細胞減少癥是一組較常見的病,這些患兒可能同時有IgM增高,遲鈍白細胞綜合征(lazyleucocytesyndrome)患兒均有中性粒細胞減少,反復上呼吸道感染,牙齦炎,口腔炎等,患者白細胞在體外試驗中,不能向趨化性刺激物移動;皮膚窗試驗中無內聚現象;給新鮮血漿後,這種異常反應也不能改變.
白細胞減少癥的病因多為遺傳性疾病,或是可以查出傢族性病史,周期性中性粒細胞減少癥則是一種骨髓幹細胞的缺陷,在中性粒細胞減少期間,單核細胞增多,並從尿中排出的粒細胞集落刺激因子增加,這說明不是粒細胞生成激素的生成障礙.
2.吞噬細胞的功能不足
(1)慢性肉芽腫病(chronicgranulomatousdisease,CGD):該病主要見於男性患兒,一般在出生後數年內,反復發生化膿性感染,癤及蜂窩織炎最為常見,常並發淋巴結化膿,進而引起骨髓炎,這類患者的白細胞吞噬細菌後,卻不能把它們殺死,反而受到白細胞的保護,不受其抗體,補體及藥物的影響.
(2)Chédiak-Higashi綜合征:這是一種少見的綜合征,患兒多形核白細胞數降低,並缺乏正常功能,對趨化性刺激反應不良,殺菌能力低下,患者反復發生嚴重的化膿性感染,伴有皮膚色素缺乏,羞明,肝脾腫大等,能存活至成人者極少.
(3)Job綜合征:是累及女孩的吞噬功能缺陷病,主要表現為不能清除入侵的葡萄球菌,患兒伴有色素缺乏和濕疹.
上述遲鈍白細胞綜合征,屬於白細胞功能缺陷,其他趨化性缺陷,可能檢出補體缺乏或白細胞抗體等.

吞噬功能缺陷病
吞噬功能缺陷病症状
1.慢性肉芽腫病(chronicgranulomatousdisease,CGD),大多在幼年發病,常有顯著的淋巴結腫大,肝脾腫大,深部膿腫和肺炎,致病菌多為過氧化物酶陰性者,如表皮葡萄球菌,金黃色葡萄球菌,沙雷菌,大腸埃希桿菌和假單胞菌等,其他癥狀有鼻炎,結膜炎,皮炎,肝膿腫等,病人一般發育延遲.
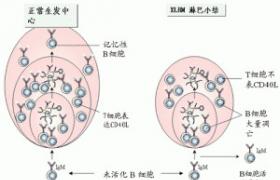
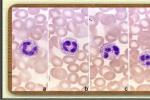
2.Chédiak-Higashi綜合征,本病為一罕見的累及多臟器的常染色體隱性基因的遺傳病,臨床上表現為:反復性化膿菌感染,部分性眼和皮膚白化病,中樞神經系統異常,臟器有非惡性的淋巴細胞浸潤,很多組織細胞內的溶酶體異常,導致感染(中性粒細胞),白化病(黑色素細胞),神經系統疾病(神經細胞)和出血(血小板).
吞噬功能缺陷病
吞噬功能缺陷病检查
1.慢性肉芽腫病實驗室檢查
白細胞計數因感染而可能增高,但可能由於NADP氧化酶活性缺乏而不能產生過氧化氫,超氧化物和其他活性氧,其他粒細胞蛋白可能也缺乏,粒細胞四唑氮藍(NBT)還原試驗缺失,殺菌試驗異常.
2.Chédiak-Higashi綜合征
實驗室檢查:患者中性粒細胞減少,趨化和殺菌力減弱,NK細胞活性降低,但殺傷T細胞活性則正常.
Chédiak-Higashi綜合征,在光學顯微鏡下可見中性粒細胞,單核細胞,淋巴細胞的胞質內和血小板內有較大的顆粒,此系異常的溶酶體融合而成.
吞噬功能缺陷病预防
1.預防遺傳性免疫缺陷
免疫缺陷的遺傳因素占很大比重,尤其以嚴重者為最,因此,對有免疫缺陷傾向者,如反復罹患多發性化膿癥者,無明顯傳染因子的腹瀉者,經常施用抗生素抗感染者等,在婚前,應作免疫學檢查,提請免疫實驗室進行免疫細胞,血清免疫因子及有關的細胞因子檢測,以及體外,體內免疫功能檢驗,同時,要作遺傳查詢,包括男女雙方的個人既往史,傢族史,畸形體等,對有腭裂,唇裂者可進一步檢查胸腺和胸腺功能;皮膚白化者要檢查其與Wiskott-Aldrich綜合征的關系.
2.預防致畸性的嬰兒免疫缺陷
為避免胎兒致畸引起的免疫缺陷,除如上查詢胎兒父母的個人既往史,傢族史,畸形體等外,還要避免母親在孕期受風疹病毒,巨細胞病毒等感染,防止服用有致畸傾向的藥物,防止有害射線,如γ射線,X射線輻照等等,產前檢查中,要註意胎兒有否畸形,畸形兒可以中斷妊娠.
3.預防繼發性免疫缺陷
加強體育鍛煉,保持身心健康,防止過度疲勞和營養不良,積極治療可能導致免疫缺陷的傳染性疾病,正確使用治療藥物,免疫抑制劑或免疫調節劑;必要時補充缺失的免疫因子,保證免疫功能正常.
吞噬功能缺陷病治疗
就診科室:內科免疫科兒科學小兒免疫科
治療方式:藥物治療支持性治療
治療周期:10-30天
治愈率:1%
常用藥品:胸腺肽腸溶片烏苯美司片
治療費用:根據不同醫院,收費標準不一致,市三甲醫院約(50000--100000元)
(一)治療
1.慢性肉芽腫病,根據不同的致病菌給予積極的抗菌治療.重組IFN-γ可促進細胞色素的轉錄,使細胞內超氧陰離子水平增加,初步結果有效.嚴重病例可予骨髓移植.
2.Chédiak-Higashi綜合征以抗生素治療感染為主,其他無特殊療法.
(二)預後
1.慢性肉芽腫病預後取決於是否及時診斷和治療,一般預後差,很少能存活至20歲以上.
2.Chédiak-Higashi綜合征,預後不良,多數在兒童期死亡,並發淋巴網狀系統惡性腫瘤的頻率增高.
吞噬功能缺陷病饮食
具體飲食建議需要根據癥狀咨詢醫生,合理膳食,保證營養全面而均衡.
飲食宜清淡,戒除煙酒,禁食辛辣刺激性食物.
吞噬功能缺陷病并发症
1.慢性肉芽腫病可並發肝脾腫大,深部膿腫和肺炎,其他還有鼻炎,結膜炎,皮炎,肝膿腫等.
2.Chédiak-Higashi綜合征,可並發白化病(黑色素細胞),神經系統疾病(神經細胞)和出血(血小板).
1/2 1 2 下一页 尾页


















