小兒遺傳性慢性進行性腎炎
小兒遺傳性慢性進行性腎炎百科
遺傳性腎炎又稱Alportssyndrome(AS),屬一種傢族性慢性進行性腎炎,臨床特征以血尿為主,部分病例可表現為蛋白尿或腎病綜合征.常伴有神經性聽力障礙及進行性腎功能減退.且近10年來隨著分子生物學的迅猛發展,對於AS的研究已進入分子水平和基因水平,現今在其遺傳方式、臨床表現、病理特點方面的認識已比較清楚.
小兒遺傳性慢性進行性腎炎
小兒遺傳性慢性進行性腎炎病因
一、發病原因病變的根本原因為基底膜的重要組分Ⅳ型膠原不同α鏈(α1-αa6、的突變,其編碼基因稱為COL4A1-COL4A6,分別位於不同的染色體上,約85%AS病人為性連鎖顯性遺傳(X-LinkedAS,XLAS、,致病基因定位於X染色體長臂中段Xq22,為編碼Ⅳ型膠原α5鏈(COL4A5、基因突變所致.其餘病人為常染色體隱性遺傳(autosomalrecessiveAS,ARAS、和常染色體顯性遺傳AS(autosomaldominantAS,ADAS、,前者致病基因為COL4A3或COL4A4基因,而後者具有遺傳多源性,運用免疫組織化學技術,檢測腎組織,皮膚組織中Ⅳ型膠原不同α鏈的表達,可幫助臨床上診斷此病及確定遺傳方式,另一方面,人們不僅確定瞭Alport綜合征的致病基因,且已通過各種方法檢測出瞭300餘種XLAS和數種ARAS的突變基因,並開始逐步探討患者基因型與表現型之間的關系.
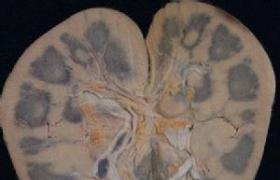
二、發病機制遺傳性慢性進行性腎炎是一種以血尿,進行性腎臟損害為主要表現的腎小球基底膜(basementmembrane,BM、病,可伴有眼,耳等腎外表現,電鏡下,基底膜表現為彌漫性厚薄不均,可有分層現象,此為特征性的病理改變,然而在發病機制方面,從Ⅳ型膠原編碼基因上單個或多個堿基突變,直至BM上的相應病變,由於缺乏動態的,系統的研究,人們尚不清楚這其中的許多具體環節,目前正在開展有關的動物模型的建立和研究,早期腎臟體積正常或增大,病情進展後腎臟體積逐漸縮小,在光鏡下腎小球正常或輕度上皮細胞增生及系膜基質增加,晚期系膜細胞增生,腎小球和腎小管基膜增厚,分層,包囊壁增厚並發展到腎小球硬化,腎小管細胞萎縮或伴部分擴張,可有蛋白管型間質可有灶性炎癥細胞浸潤,也可發展到纖維化,40%病例在皮髓質交界處的間質中有泡沫細胞浸潤,此種泡沫細胞的胞漿含有中性脂肪,黏多糖,膽固醇及磷脂,電子顯微鏡檢查典型者為腎小球及腎小管基膜變薄及不規則增厚,腎小球增厚的基膜伴致密層分裂,或重疊板層樣改變,其間含有電子致密顆粒,近年來發現腎小球基底膜(GBM、有變薄,增厚及兩者相間病變,變薄的GBM可達正常厚度的1/4,而增厚的GBM可達正常厚度的2~5倍,為節段性變薄與增厚的GBM並存,腎小球上皮部分足突可以融合或伴微絨毛形成,免疫熒光檢查通常為陰性,偶爾也能見到某些免疫球蛋白如IgM,補體C3等在腎小球內輕度沉積;一般認為系對病變腎小球內的非特異性黏附,無致病意義.
小兒遺傳性慢性進行性腎炎
小兒遺傳性慢性進行性腎炎症状
1.腎臟表現:多數以持續性或間歇性血尿為主要表現,血尿為腎小球性,也可表現程度不等的蛋白尿以及腎病樣蛋白尿,常於急性感染時加劇,受累男孩的發病可早在出生後第1年,血壓升高的發生率和嚴重性,隨年齡而增加,且多發生於男孩,受累男孩腎臟幾乎全部將發展至終末期腎臟病,但進展速度各傢系之間有差異,根據終末期腎功能衰竭的發生年齡,可分為青少年型(31歲前發生)和成年型(31歲以後).
2.神經性耳聾:約30%~40%患者可伴有高頻區(4000~8000Hz)神經性耳聾,隨著年齡的增長,小兒於學齡期逐漸出現以上癥狀,男性尤多見,兩側耳聾程度可以不完全對稱,但為進行性的,耳聾將漸及全音域,腦幹電測聽顯示聽力障礙發生於耳蝸部位.
3.眼病變:約15%患者合並有眼病變,最具特征性的眼部異常為前圓錐形晶狀體,即晶狀體中央部位突向前房,確認這一病變常需經眼科裂隙燈檢查,其他常見的眼部異常為黃斑周圍色素改變,在黃斑區中心凹周圍有致密微粒沉著,先天性白內障,眼球震顫等.
4.血液系統異常:與腎臟病相關的巨血小板減少癥已有報道,此癥患者血塗片血小板計數多在(30~70)×109/L,血小板呈球形,臨床可表現有輕度出血傾向,但極少發生術後嚴重出血現象,同時外周血塗片還可見粒細胞,甚至巨噬細胞內包涵體.
5.彌漫性平滑肌瘤:某些青少年型Alport綜合征傢系或患者伴有顯著的平滑肌肥大,受累部位常為食管,氣管和女性生殖道(如陰蒂,大陰唇及子宮等),出現相應的癥狀,如吞咽困難,呼吸困難等.
小兒遺傳性慢性進行性腎炎
小兒遺傳性慢性進行性腎炎检查
尿液檢查:血尿為持續性鏡下血尿,上感或勞累後可出現陣發性肉眼血尿,雜合子可為間歇性血尿,早期有微量蛋白尿,以後可發展至腎病樣蛋白尿,往往提示預後不良.
血液檢查:血小板巨大,數量減少,常在(30~70)×109/L,呈球形直徑,約5~15?m(正常1~2?m),另外血液塗片可見白細胞包涵體,可有尿素,肌酐升高.
腎功能檢查:在幼童期大多正常,之後男性患者腎功能逐漸減退,多數在20~30歲出現腎功能衰竭,約占小兒腎功能衰竭的3%,偶爾女性患者在青春期即進入腎功能不全.
腎活檢:
光鏡:無特殊意義的病理變化,可見有腎小管間質炎性改變,灶性毛細血管壁硬化,系膜區輕度不規則增寬,局灶性包囊增厚,局灶性內皮或系膜細胞增生等,一般5歲以前患兒的腎小球和血管正常或基本正常,可能發現的異常是皮質腎小球呈嬰兒樣腎小球改變.
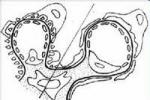
電鏡:電鏡檢查是惟一有診斷價值的方法,典型病變為腎小球基底膜(glomerularbasementmembrane,GBM)彌漫性增厚,致密層撕裂,但也可見GBM變薄,此病變改變不是Alport綜合征特有的病變,電鏡下可見全部或部分GBM致密層增厚,可達1200nm(正常為100~350nm),並有不規則的內,外輪廓線,不均一的致密層中還可見直徑約為20~90nm的電子致密物沉積,GBM撕裂,扭曲及密度不均,在年幼兒,婦女或疾病早期,GBM彌漫性變薄,常規做X線檢查,B超,腦電圖,心電圖檢查,做眼底檢查,電測聽檢查等,電測聽器檢查可發現,雙側性神經性耳聾,裂隙燈檢查發現白內障,角膜色素沉著等異常,X線骨片見肢端骨質溶解病變及食道,支氣管,生殖器平滑肌瘤.
小兒遺傳性慢性進行性腎炎预防
本癥屬一種傢族性慢性進行性腎炎,約85%遺傳性慢性進行性腎炎病人為性連鎖顯性遺傳,為基因突變所致;其餘病人為常染色體隱性遺傳和常染色體顯性遺傳,前者致病基因為COL4A3或COL4A4基因,而後者具有遺傳多源性,因此,應詳細進行傢系調查,做好遺傳咨詢,進行生育指導,遺傳性疾病是影響嬰兒和兒童健康的重要原因,影響出生人口的素質,為降低和扭轉出生缺陷的發生率,預防應從孕前貫穿至產前:
婚前體檢在預防遺傳性疾病中起到積極的作用:作用大小取決於檢查項目和內容,主要包括血清學檢查(如乙肝病毒,梅毒螺旋體,艾滋病病毒),生殖系統檢查(如篩查宮頸炎癥),普通體檢(如血壓,心電圖)以及詢問疾病傢族史,個人既往病史等,做好遺傳病咨詢工作.
孕婦盡可能避免危害因素:包括遠離煙霧,酒精,藥物,輻射,農藥,噪音,揮發性有害氣體,有毒有害重金屬等,在妊娠期產前保健的過程中需要進行系統的出生缺陷篩查,包括定期的超聲檢查,血清學篩查等,必要時還要進行染色體檢查,一旦出現異常結果,需要明確是否要終止妊娠;胎兒在宮內的安危;出生後是否存在後遺癥,是否可治療,預後如何等等,采取切實可行的診治措施.
小兒遺傳性慢性進行性腎炎治疗
(一)治療
遺傳性慢性進行性腎炎為先天性遺傳性疾病,至今尚無特效治療.
1.一般治療:以對癥,控制並發癥為主,積極預防繼發性尿路感染,防止過度疲勞及劇烈體育運動,遇有感染時,避免應用腎毒性藥物,動物試驗表明,嚴格控制蛋白,脂肪,鈣,磷攝取,對延緩腎功能減退和抑制腎小球病變的進展有幫助.
2.終末期腎功能衰竭階段:則依靠透析療法維持生命,等待腎移植,由於Alport綜合征病人體內缺乏基膜抗原,在腎移植後可產生抗GBM的抗體,以致移植腎發生抗GBM腎炎(Goodpasture綜合征),故有作者主張對這些病人進行腎移植後,應密切追蹤尿常規,腎功能及血清抗GBM抗體,至少追蹤1年.
3.基因治療:部分Alport基因已被確定,使下一步基因治療成為可能,但真正實施基因治療尚有待時日.
(二)預後
遺傳性慢性進行性腎炎的預後與其基因突變的特點,遺傳方式有關,反復血尿可持續多年,其預後與性別密切相關,男性患者通常於20歲後漸進入慢性腎功能衰竭,據前述EDTA的資料,479例男性患者平均24.3歲時,進入慢性腎功能衰竭,需行替代療法,女性患者較輕,很少進入腎衰,據我國77例材料,48例男性患者中,25例(52%)死於腎功能衰竭,其中91.2%於20歲前已死亡,女性患者29例中有6例死於本癥.
小兒遺傳性慢性進行性腎炎饮食
飲食方面要做到規律、合理,即以高蛋白、高維生素食物為主.選擇營養價值高的植物或動物蛋白,如牛奶、蛋類、魚類、瘦肉、各種豆制品等.
小兒遺傳性慢性進行性腎炎并发症
此外疾病後期發生高血壓、腎功能進行性減退,發展為腎功能衰竭.同時有斜視、近視、視力減退、白內障.神經性耳聾也呈進行性加重.此外疾病後期發生高血壓、腎功能進行性減退,發展為腎功能衰竭.同時有斜視、近視、視力減退、白內障.神經性耳聾也呈進行性加重.
1/2 1 2 下一页 尾页


















