進行性肌營養不良癥 進行性肌營養不良
進行性肌營養不良癥 進行性肌營養不良百科
進行性肌營養不良癥(progressivemyodystrophy)是一組由遺傳因素所致的原發性骨骼肌疾病,其臨床主要表現為緩慢進行的肌肉萎縮,肌無力及不同程度的運動障礙.本病可由多種遺傳方式引起,其臨床表現各具有不同的特點,因而形成許多類型.本病可由多種遺傳方式引起,多發生於兒童和青少年.

進行性肌營養不良癥 進行性肌營養不良
進行性肌營養不良癥 進行性肌營養不良病因
(一)發病原因
本病的發病機制研究已為世人所矚目,數十年來,相繼提出的有血管性,神經性,肌纖維再生錯亂和細胞膜缺陷等學說,但大量的研究證據表明細胞膜缺陷在本病發生有重要地位,三分之一新生男嬰患兒是由於基因突變所引起.
隨著分子生物學研究的深入開展,本病的病因和發病機制有瞭進一步闡明,已明確本病是一類單基因遺傳病,遺傳方式多樣,不少致病基因已得到定位和克隆,部分基因產物已得到闡明,有的致病基因尚不明,相關基因位點突變可引起其表達產物肌膜結構蛋白的缺陷和異常而致病.
對其不同類型,不同亞型的分子機制也有瞭新的認識,其中以Duchenne型和Becker型肌營養不良(DMD,BMD)的研究最為深入,DMD為X連鎖隱性遺傳病,致病基因已定位於X染色體短臂2區1帶2~3亞帶(Xp21.2~21.3),其基因的cDNA已經被克隆,全長為14kb,有60~65個外顯子,基因表達產物為抗肌萎縮蛋白(dystrophin,Dys),當基因出現大片段缺失,重復或其他形式變異如點突變時,導致Dys缺如或結構功能異常是DMD致病的根本原因,BMD基因與DMD處於同一區域,互為等位基因,Dys位於肌纖維膜的內層,是一種細胞骨架蛋白,有穩定肌纖維膜的作用,在DMD患者中,由於肌纖維缺乏Dys,肌膜結構的完整性受到破壞,大量富含鈣離子的細胞外成分流入肌細胞內,最終導致肌纖維變性,壞死而發病,Emery-Dreifuss肌營養不良的致病基因定位於xq28,其編碼蛋白為emerin,近年來發現肢帶型肌營養不良(LGMD)的發生與附著於肌纖維膜上的抗肌萎縮蛋白-糖蛋白復合物(dystrophin-glycoproteincomplex,DGC)的遺傳缺陷有關,DGC在維持肌纖維膜的穩定和防止膜損傷壞死起著十分重要的作用,面肩肱型肌營養不良(FSHD)為成年人最常見的常染體顯性遺傳肌病,其基因定位存4q35,基因尚未克隆,編碼蛋白尚未分離,但已證明FSHD與4號染色體長臂末端3.3kb串聯重復序列拷貝數的缺失有關,其他如遠端型肌營養不良不同亞型的分子機制也有新的認識.
(二)發病機制
與肌營養不良發病有關的膜結構蛋白是由多種蛋白質組成的一個大復合物,稱抗肌萎縮蛋白-糖蛋白復合物(dystrophin-glucoproteincomplex,DGC),包括抗肌萎縮蛋白(dystrophin),肌營養不良聚糖復合物(由α,β-dystroglycan組成),肌聚糖復合物(α,β,γ,δ-sarcoglycan)和營養合成蛋白復合物(syntrophincomplex),dystrophin的一端與肌動蛋白相聯結,另一端與β-dystroglycan相聯結,再通過α-dystroglycan與基底膜上的細胞外基質蛋白α2-Laminin相聯結,在結構上起到瞭連接肌肉細胞內肌動蛋白和細胞外基質的橋梁作用,DGC的各組成分緊密結合,相互關聯可維護肌膜的穩定性和完整性,當相應的基因位點發生突變,使DGC某一組份發生缺陷時,如dystrophin或任一型sarcoglycan的缺乏,都會影響整個膜結構的穩定,引起肌膜損傷,進而發生一連串反應並導致肌纖維壞死.
本病早期病理變化僅見肌纖維大小不等,內核增多,病變進展期表現肌纖維結構紊亂,大小懸殊明顯,在同一肌束中萎縮纖維,撕裂纖維及肥大纖維呈不規則的混雜分佈,光鏡下見肌纖維粗細不等,肌纖維變性,壞死,如玻璃樣變性,顆粒狀變性,絮狀變性及吞噬現象等,肌膜核內移,排列成鏈狀,早期可見再生纖維,晚期肌纖維消失殆盡,由脂肪及結締組織所代替.
進行性肌營養不良癥 進行性肌營養不良
進行性肌營養不良癥 進行性肌營養不良症状
傳統分為以下類型:
1.假性肥大型肌營養不良(pseudohypertrophicmusculardystrophy)
X性連鎖隱性遺傳,基因位點在Xp21,基因的缺陷可導致骨骼肌中其編碼蛋白dystrophin的缺乏,分為Duchenne和Becker兩型,前者起病年齡早,病情重,進展快,dystrophin幾乎缺如;後者起病年齡較遲,病情相對較良性,dystrophin量減少或有質的改變.
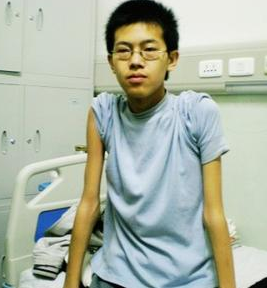
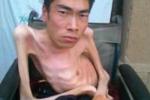
(1)Duxhenne型(Duchennemusculardystrophy,DMD):是肌營養不良中發病率最高,病情最為嚴重的一型,常早年致殘並導致死亡,故稱為“嚴重型",幾乎所有患者均為男孩,女孩患病極為罕見,多在3歲之後發病,可見患兒動作笨拙,跑,跳等均不及同齡小孩,因骨盆帶及股四頭肌等無力,致使行走緩慢,易跌倒,登樓上坡困難,下蹲或跌倒後起立費勁;站立時腰椎過度前凸,步行時挺腹和骨盆擺動呈“鴨步"樣步態,仰臥起立時,必須先翻身與俯臥,以雙手撐地再扶撐於雙膝上,然後慢慢起立,稱Gower征,隨病情發展累及肩帶及上臂肌時,則雙臂上舉無力,呈翼狀肩胛,萎縮無力的肌肉呈進行性加重,並可波及肋間肌等,假性肌肥大最常見於雙側腓腸肌,因肌纖維被結締組織和脂肪所取代,變得肥大而堅硬,假肥大也可見於三角肌,股四頭肌等其他部位的肌肉,肌腱反射減弱或消失,隨肌萎縮無力之加重及關節活動的減少,可出現肌腱攣縮及關節強硬畸形,大約在12歲左右便不能站立和行走,不少患兒伴心肌病變,心電圖多有異常,如高R波,Q波加深等,部分患兒智力低下,大約在20歲左右,病人多因呼吸衰竭,肺部感染及心力衰竭等原因而死亡.
(2)Becker型(Bekermusculardystrophy,BMD):與DMD相似,區別要點主要在於病程長,發展相對緩慢,有一段正常的生活期,故稱之為“良性型",本型一般在5~20歲發病,大約在出現癥狀後20餘年才不能行走,四肢近端肌肉萎縮無力,尤以下肢明顯,腓腸肌肥大常為早期征象,心肌受損及關節攣縮畸形較少見,智力一般正常,大多可存活至40~50歲.
2.Emery-Dreifuss肌營養不良
是一種少見的良性X連鎖隱性遺傳病,多於2~10歲發病,初期常表現上肢近端及肩胛帶肌無力,數年後逐漸累及骨盆帶及下肢遠端肌群,一般以脛骨前肌和腓骨肌無力和萎縮最為明顯,少數可伴有面肌輕度無力,本型常在早期出現頸,肘,膝,踝關節攣縮,幾乎所有病人均伴有不同程度的心臟損害,可由心臟傳導阻滯而突然致死.
3.面肩肱型肌營養不良(facioscapulohumeralmusculardystrophy,FSHD)
為常染色體顯性遺傳病,男女均可罹患,發病年齡差異很大,一般為5~20歲.
病變主要侵犯面肌,肩胛帶及上臂肌群,面肌受累時表現面部表情淡漠,閉眼,示齒力弱,不能蹙眉,皺額,鼓氣,吹哨等,由於常合並口輪匝肌的假性肥大,以致上下嘴唇增厚而微噘,同時病變會延及雙側肩胛帶及臂肌群,常為不對稱性,以致患者雙臂不能上舉,外展不能過頭,出現梳頭,洗臉,穿衣等困難,由於肩胛帶肌無力萎縮,表現明顯的翼狀肩,有的表現遊離肩或“衣架樣肩胛",可見三角肌,腓腸肌假性肥大,心肌受累罕見,晚期才累及骨盆帶肌群,病情進展緩慢,一般預後較好.
4.肢帶型肌營養不良(limbgirdlemusculardystrophy,LGMD)
以往由於對該類病變認識甚少,隻是根據臨床癥狀和遺傳方式來分型的,隨著分子生物學研究的深入,Bushby和Beckmann(1995)根據基因分析的結果,對LGMD提出一個全新的分型命名,他們按遺傳方式將LGMD分為兩型:LGMD1代表常染色顯性遺傳,LGMD2代表常染色體隱性遺傳;並在LGMD1或LGMD2後加字母表示不同的致病基因所導致的相應亞型,截止目前,LGMD1分為LGMD1A,1B和1C3種類型;LGMD2則分LGMD2A,2B,2C,2D,2E,2F,2G和2H,共8種類型,在LGMD中,90%以上為LGMD2.
現將其中較常見的類型簡述如下:
(1)LGMD1A型:基因定位於5q22.3-q31.3,其編碼蛋白為myotilin,多在青壯年期間發病,初期表現為四肢近端無力,逐漸累及肢體遠端,後期見有踝關節攣縮,病情進展緩慢,最終失去行走能力,血清CPK水平升高,EMG呈肌源性損害.
(2)LGMD2A:基因定位於15q15.1-p121.1,其編碼蛋白為calpain-3,臨床嚴重程度不一,大部分表現較輕,發病年齡4~15歲,主要表現為雙下肢近端無力,呈對稱性,後累及肩胛帶肌群,多於30歲左右喪失行走能力,有些患者可有腓腸肌假性肥大,但程度較輕,後期可有小腿肌攣縮,脊柱強直,血清CPK水平明顯升高.
(3)LGMD2C(重型兒童常染色體隱性遺傳性肌營養不良,SCARMD):基因定位於13q12,編碼蛋白為r-sarcoglycan,病情嚴重,部分病例有類似DMD的病程,其他多介於DMD和BMD之間,發病年齡為3~12歲,首先侵犯骨盆帶肌,以後波及胸部,頸部肌,尚伴有心肌受累,一般不影響智力,多有腓腸肌假性肥大,常於10~13歲喪失行走能力,30~40歲出現呼吸衰竭,血清CPK水平明顯升高.
5.眼咽型肌營養不良(oculopharyngealmusculardystrophy)
屬常染色體顯性遺傳性肌病,多在40歲左右起病,首先出現對稱性眼外肌無力和(或)眼瞼下垂,後逐漸表現吞咽,構音困難,進展十分緩慢,少數患者以吞咽障礙作為首發癥狀,尚有些患者伴有輕度的面肌,咬肌,顳肌以及肢帶肌等的無力和萎縮.
6.遠端型肌營養不良(distalmusculardystrophy)
目前已將該型肌營養不良至少分為4個亞型,即常染色體顯性遺傳Ⅰ型,Ⅱ型及常染色體隱性遺傳Ⅰ型,Ⅱ型,前者多出現在歐洲,而日本報道的病例多為常染色體隱性遺傳Ⅰ型和Ⅱ型,該類肌病的共同特點是:肌無力主要表現在四肢的遠端,以伸肌的無力和萎縮最明顯;無感覺障礙及自主神經損害的表現;肌電圖為肌源性損害,其中有些類型的病理學檢查與遺傳性包涵體肌病相似.
7.強直性肌營養不良(myotonicdystrophy)
本病為常染色體隱性遺傳,致病基因定位於19q13.3,編碼蛋白為強直性肌營養不良蛋白激酶(myotonicdystrophyproteinkinase,MDRK)或稱DM-kinase(DMK),正常健康人的DMK有5~37個CAG核苷酸重復序列,而強直性肌營養不良患者該基因CAG重復可達50~300個,此類由於三核苷酸串重復導致的疾病統稱為三核苷酸重復疾病(tripletrepeatdiseases),本病的病理特點與其他類型的肌營養不良不同,肌纖維壞死和再生少見,而主要改變為肌纖維周邊大量的肌漿塊形成,內核肌纖維明顯增多,縱切面可見核鏈形成,此外還可有選擇性Ⅰ型纖維萎縮,因此現在有一種觀點認為強直型肌營養不良在分類上不屬於肌營養不良,而屬強直性肌病的范疇.
本病又稱營養不良性肌強直(dystrophiamyotonica),臨床分為成人型,先天型和輕癥型三種類型,發病年齡和疾病的嚴重程度有關,發病越早,臨床癥狀越重,頭面諸肌,頸肌和四肢遠端肌肉受累較重,表現為雙瞼下垂,咬肌和頰肌萎縮形成特有的“斧型臉";胸鎖乳突肌萎縮無力致使頸部彎曲,過度前傾,形成“天鵝頸",早期即可有脛骨前肌無力,萎縮和足下垂,咽喉肌受累可導致鼻音,語音單調,聲音低鈍,食管上部骨骼肌受累可引起食管擴張,隨病情進展,近端肌群和骨骼肌也受累,腱反射低下或消失,肌強直表現為輕輕叩擊或電刺激後,肌肉出現自發性長時間收縮,大魚際肌,舌肌和眼輪匝肌容易誘發,強直癥狀可先於肌肉無力多年出現,有些患者早期可能被誤診為先天性肌強直,先天型和嬰兒期發病的強直型肌營養不良,早期較長的一段時間內可無肌強直癥狀,有些甚至在20~30歲以後才出現,本病多在15~20歲喪失行走能力,多數患者不能存活到正常壽命.
強直型肌營養不良為多系統損害疾病,除肌萎縮,肌無力和肌強直外,還有內分泌系統損害如陽痿,脫發,睪丸萎縮,乳房腫大和卵巢功能下降;心臟損害如心律失常,房室傳導阻滯;神經精神損害如精神發育遲滯,遺忘,多疑;眼部損害如晶體渾濁和白內障(見於90%的患者),有些患者還可以伴有運動感覺性周圍神經病.

進行性肌營養不良癥 進行性肌營養不良
進行性肌營養不良癥 進行性肌營養不良检查
1.血生化檢查
血清肌酸磷酸激酶(CPK)增高是重要而敏感的指標,以假性肥大型升高最明顯,肢帶型次之,面肩肱型輕度升高或正常,在假性肥大型的早期CPK增高最為顯著,晚期活性下降,此外,血清肌紅蛋白(Mb),丙酮酸激酶(PK)及乳酸脫氫酶(LDH)也是較敏感的指標,丙氨酸氨基轉移酶(ALT)和天門冬氨酸氨基轉移酶(AST)也常升高,多種酶指標的聯合測定更有利於相互參照.
2.尿肌酸測定
24h尿液肌酸排出量增高.
3.肌電圖
松弛時可出現自發電位,輕收縮時運動單位電位的平均時限縮短,平均波幅降低,出現短棘波多相電位,強收縮時呈病理幹擾相,峰值電壓一般小於1000μV.
4.骨骼肌CT或MRI檢查
通過多部位骨骼肌的CT或MRI影像檢查可瞭解骨骼肌損害的分佈范圍和嚴重程度,有助於早期診斷和提供肌肉活檢的優選部位.
5.肌活檢
(1)形態學:光鏡和電鏡下顯示骨骼肌的病理改變見前所述.
(2)骨骼肌基因產物--蛋白的測定:以相應蛋白的特異性抗體,應用免疫組化技術和免疫印跡技術檢測骨骼肌中相應蛋白質的分佈以及其質和量的變化,如Duchenne型肌營養不良的骨骼肌膜dystrophin幾乎缺如.
6.心功能檢查
90%DMD患者伴有心臟損害,一般心電圖檢查多可出現竇性心動過速,異常R波,V1導聯S波變淺,深的Q波,P-R間期縮短以及束支傳導阻滯等異常,Emery-Dreifuss肌營養不良在心功能檢查方面常有心肌損害,心律失常和心臟傳導障礙等異常表現,而在其他類型心臟受累均較少見.
7.基因檢測
采取病人外周血,運用分子生物學技術,對致病基因進行直接檢測或間接進行連鎖分析,從DNA水平上進行診斷,如在Duchenne肌營養不良中檢測外顯子的缺失或其他類型的基因缺陷.
(1)DMD/BMD基因檢測:在DMD基因缺陷中,有65%為缺失突變,5%為重復突變,其餘為點突變及其他突變形式,目前,可針對其不同的突變形式采取不同的方法進行診斷:
①對於基因缺失和重復者,可聯合應用多對引物進行多重PCR擴增.
②對於非缺失型,多采用PCR-STR連鎖分析法.
③對於點突變者,可采用PCR-SSCP和DNA測序技術.
(2)FSHD基因檢測:近年來研究發現,95%以上的FSHD病例與4q35區的3.3kb重復單位缺失,導致該區的一個EcoRI片段縮短有關,此片段可通過P13E-11探針經Southern雜交的方法進行檢測,正常人此片段為35~300kb,患者由於上述缺失而小於35kb,因此,直接檢測該片段大小可對本病進行基因診斷.
進行性肌營養不良癥 進行性肌營養不良预防
預防本病的惟一有效手段是遺傳咨詢,產前診斷和選擇性流產,特別對DMD/BMD,可采用生化方法,如血清CPK,Mb檢測,有助於判定致病基因攜帶者,分子生物學技術的應用,如cDNA探針檢測,PCR擴增,Dys印跡及免疫熒光檢查等,大大提高瞭DMD/BMD致病基因攜帶者的檢出率,並可用於產前基因診斷,這對控制本病的發生具有極重要的意義.
進行性肌營養不良癥 進行性肌營養不良治疗
(一)治療
本病目前尚無特效的治療方法.隻能采取對癥療法及一般支持療法,包括應用維生素E、肌苷、加蘭他敏、三磷腺苷、苯丙酸諾龍以及中藥等.適當的功能鍛煉,進行各關節充分被動運動,針灸、推拿、按摩等均可延緩更嚴重的肌無力、肌萎縮和關節攣縮的發生.
積極預防和治療呼吸道感染,對延長患者的存活時間是有價值的.國外報道采用皮質類固醇作為DMD的治療,對改善患者的肌力和運動功能,延緩病程的進展有一定作用.但長期應用這類藥物副作用較大,且其遠期療效如何.還需作進一步觀察.
有關DMD的基因治療,目前還限於動物試驗階段.由於Dys基因是迄今人類發現的最大基因之一,介導全長14kbcDNA進入肌肉細胞尚難以實施.以往采用的病毒或非病毒轉基因系統,都存在著轉移效率低以及其他問題.新近匹茲堡大學的研究人員構建出一種小於4.2kb微型Dys基因,可裝載入腺病毒相關病毒載體,導入mdx鼠肌細胞,該轉移系統可長期維持具有治療意義的Dys蛋白表達,這是DMD基因治療方面最為引人註目的進展.幹細胞是一種能分化為多種組織細胞的始祖細胞,有報道靜脈註射正常造血幹細胞,可使mdx大鼠造血功能重建,並部分恢復受累肌細胞Dys的表達,因而幹細胞移植在近年來也成為DMD治療研究的又一熱點.
(二)預後
不同類型,預後不同.參見臨床表現,此處不贅述.
進行性肌營養不良癥 進行性肌營養不良饮食
1、飲食宜清淡、營養豐富,忌食或少食油膩厚味過熱、傷津耗液及損傷脾胃之品,可多食魚類、蛋類、雞肉、瘦豬肉等,但不可太過,以免損傷脾胃.白菜、豆芽、西紅柿、山楂、廣柑、棗子之類的蔬菜水果可以適當多食一些.
2、堅持體育鍛煉,自我按摩以增加活動,促進血液循環,防止肌肉萎縮,但應適度,不可過勞.積極與疾病作鬥爭,堅持適當的娛樂活動,促使患者建立樂觀、開朗的情緒,樹立以堅強毅力戰勝疾病的信心.
3、采用力所能及的鍛煉,亦不要過勞:上肢可練習抬舉、俯臥撐、擴胸等;腰部可練習仰臥起坐;下肢可練習起蹲、上樓、跳躍、側壓腿等;註意防止攣縮,對膝關節、跟腱關節熱敷後適當牽引;假肥大部位的按摩以揉法為主;防止脊柱畸形,保持良好坐姿,勞累後宜平臥休息.
進行性肌營養不良癥 進行性肌營養不良并发症
晚期,四肢攣縮,活動完全不能.常因伴發肺部感染,褥瘡等於20歲之前喪生.智商常有不同程度減退,半數以上可伴心臟損害,心電圖異常.早期呈現心肌肥大,除心悸外一般無癥狀.
1/2 1 2 下一页 尾页


















