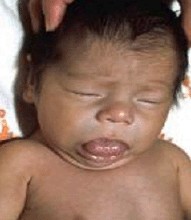
生長激素缺乏癥 E23.013 GHD
生長激素缺乏癥 E23.013 GHD百科
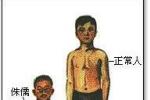
生長激素(GH)是人體促進生長的主要因素.影響身高的因素很多,在兒童、青少年時期,垂體GH分泌不足或缺乏,患者的生長都會出現明顯障礙.其他原因還有營養不良、種族遺傳、社會心理因素、多種軀體疾病包括內分泌與非內分泌性疾病等.如果身高低於同民族、同年齡的30%以下,或成人身高小於120cm,就是侏儒癥(dwarfism).
生長激素缺乏癥 E23.013 GHD
生長激素缺乏癥 E23.013 GHD病因
一)發病原因
特發性生長激素缺乏癥
占此類病人的絕大多數,其具體原因尚不十分清楚,現有的資料表明,大多數特發性生長激素缺乏癥的原因病變在下丘腦,其生長激素釋放激素(GHRH)的釋放明顯減少,一些屍檢資料發現,垂體前葉內有足夠的生長激素細胞數目與相當的細胞內生長激素儲存,這些患兒對用GHRH及其類似物治療的反應良好,GHRH不僅能促使垂體的生長激素細胞生長發育與分泌激素,它還能促使GH基因的表達,從而促進GH基因的轉錄,翻譯,合成與儲備,現在較普遍認為此癥是患兒有早期下丘腦病變,使GHRH細胞受損,最終GHRH的分泌頻率,分泌量均減少,使垂體的GH細胞接收不到足夠的信號,以致GH細胞發育不良,激素的儲存與分泌減少,導致下丘腦GHRH細胞受損的直接原因,現在看來是多因素的,包括出生前下丘腦發育不良,出生時和出生後的感染,甚至與自身免疫有關,還可能與出生時的缺氧,產傷有關,這些因素完全有可能引起其他下丘腦和垂體的激素分泌異常,但特發性生長激素缺乏癥幾乎都是孤立的生長激素缺乏,該類病人受致病因子作用後,如產生其他的釋放激素或釋放抑制激素,臨床與實驗室檢查在現有的激素檢測水平上應能發現;看來致病因子還不十分明確,上述可能的原因與GHRH細胞選擇性受損的環節還有待進一步研究,另外有相當一部分病人對GHRH治療無明顯反應或反應不良,特發性垂體性侏儒不僅有下丘腦的原發病變,垂體本身也可能因產傷,出生時感染和缺氧,以及出生後感染,自身免疫等因素引起病變,由於該病的病情是一種良性過程,目前尚缺乏直接與下丘腦和垂體有關的生理,生化,病理的詳細資料.
先天性生長激素缺乏癥
先天性GH缺乏的病人出生時身高正常,仔細檢查第1年內就可發現生長遲緩,1~2歲就有明顯的生長障礙,其GH缺乏的程度可重可輕,輕者生長也較明顯遲緩,因為這一類病人除GH缺乏外,多數還有其他下丘腦垂體激素的缺乏,患兒智力一般正常,CT或MRI可發現下丘腦和垂體部有器質性病變,也可無特殊發現;該病變部位可有或無解剖異常.
先天性垂體缺如是一種常染色體隱性遺傳病,該病非常罕見,患兒有嚴重的腺垂體功能減退,無或隻有空的蝶鞍.
遺傳性生長激素缺乏癥實際上是GH基因缺失引起的,此類病人病情多嚴重,出生後6個月就能發現生長障礙,此類病人中最常見的是GH基因中7.5kb的堿基片段丟失引起的垂體性侏儒,按遺傳方式與對治療的反應和體內內源性生長激素的活性,可分為4種.
下丘腦及垂體的器質性病變
下丘腦和垂體受到後天性破壞,可使病人出現獲得性的GH缺乏或減少,臨床上常見的情況有腫瘤(主要是顱咽管瘤),席漢(Sheehan)綜合征,外傷,感染,垂體卒中,下丘腦和垂體受過放射源照射等,此類患兒多伴有其他垂體功能不足的癥狀,單一GH缺乏很少見;即使臨床以生長障礙為主,多半還能檢查到其他激素缺乏的表現.
社會心理因素
此類病人在國內未見報道,患兒營養正常,但可有怪癖等心理障礙,垂體的GH細胞正常,GH分泌水平在一部分病人中低下,一部分正常,主要可能與GHRH的脈沖式分泌失常,導致GH的分泌方式,脈沖的頻率與時間和波幅異常有關,多認為是患兒對生長的傢庭環境不適應,有抵觸或受虧待的心理,改變生活環境,多數患兒的生長可恢復正常;此類患兒用GH或GHRH替代的療效較差.
二)發病機制
大多數患者無明顯原因可查,謂之特發性GH缺乏癥(idiopathicgrowthhormonedeficiency,IGHD),由於很多IGHD患兒有圍生期病變史(如早產,難產,窒息等),故IGHD(在有些文獻中孤立性GH缺乏癥也縮寫為IGHD)的發生可能與圍生期腦部的受損有關,IGHD患兒對單次GHRH反應不佳,但若重復給予GHRH則多數患兒血GH可升至正常,說明GH的缺乏可能繼發於GHRH的不足.
有些GH缺乏癥表現出明顯的傢族遺傳特點,謂之遺傳性或傢族性GH缺乏癥(familialgrowthhormonedeficiency),根據是否伴有其他垂體激素的缺乏,傢族性GH缺乏癥分為孤立性GH缺乏癥(isolatedgrowthhormonedeficiency)和多激素型GH缺乏癥(或稱聯合性垂體激素缺乏癥)2種,根據遺傳特點,孤立性GH缺乏癥又分為3種類型:Ⅰ型為常染色體隱性遺傳,Ⅱ型為常染色體顯性遺傳,Ⅲ型為Ⅹ-連鎖GH缺乏癥,其中Ⅰ型還分為ⅠA和ⅠB2個亞型,ⅠA亞型由GH-1基因(或稱GH-N基因)缺失所致,ⅠB亞型既可由GH-1基因突變引起,也可由GHRH受體基因突變引起,Ⅲ型的致病基因尚不清楚,同孤立性GH缺乏癥一樣,多激素型GH缺乏癥也分為3型,Ⅰ型為常染色體隱性遺傳,Ⅱ型為常染色體顯性遺傳,Ⅲ型為Ⅹ-連鎖GH缺乏癥,Ⅰ型多激素型GH缺乏癥多因Propl基因突變所致,Ⅱ型多因Pit-1基因突變所致,Ⅲ型的致病基因則不清楚.
大多數孤立性GH缺乏癥因GH-1基因突變所引起,GH-1基因位於17號染色體的長臂,GH-1基因的下遊含有GH-2基因(或稱GI-V基因)及3個CS基因,它們與GH-1基因的同源性很高,這些基因可發生不等交換,從而造成GH-1基因的缺失,GH-1基因還可發生點突變,GH-1基因的缺失可造成機體不能合成和分泌GH從而產生嚴重的ⅠA型GH缺乏(但其他垂體激素不受影響),此種病人在胎兒期即缺乏GH,不過由於胎兒的生長不依賴於GH,故患兒在宮內生長不受影響,患兒在發育過程中由於缺乏GH,因此沒有形成對人GH(hGH)的免疫耐受,他們在接受hGH治療時極易產生抗hGH抗體從而對hGH產生抵抗,有些病人GH-1基因發生點突變或重排,但病人仍能表達GH,且表達的突變GH具有一定的功能(但活性降低),為ⅠB型GH缺乏,此種病人於胚胎期已形成對hGH的免疫耐受,外源性hGH治療不易產生抗體,GHRH受體的突變使GHRH的作用減弱從而引起GH缺乏,也屬於ⅠB型GH缺乏癥,從理論上說,GHRH基因的突變亦可引起GH缺乏,但迄今尚沒有在人類GH缺乏癥中發現該基因的突變.
為什麼GH-1基因的突變既可表現為常染色體隱性遺傳(Ⅰ型),又可表現為常染色體顯性遺傳呢(Ⅱ型)?常染色體隱性GH缺乏癥的形成機制是容易理解的,因為雜合子含有一個正常的GH-1基因,其表達產物可維持正常的GH分泌,故雜合子不發病,常染色體顯性GH缺乏癥的形成機制可能與顯性負(dominantnegative)效應有關,這類病人的突變基因可能編碼一種無功能GH,此種無功能GH可與正常GH競爭GH受體,雜合子病人雖有1個正常的GH-1基因,但其表達的正常GH因突變GH的存在而失去瞭功能,這就是所謂顯性負效應,顯性負效應使得雜合子也發病,從而表現為常染色體顯性遺傳.
多激素型GH缺乏癥一般由一些在垂體發育和垂體激素基因表達中具有重要作用的轉錄因子突變所引起,Pit-1是一種垂體特異的轉錄因子,為POU傢族成員,屬螺旋-轉角-螺旋形轉錄因子,GH-1基因啟動子上遊含有Pit-1,反應元件,可結合Pit-1同二聚體(2個Pit-1單體組成的復合體),Pit-1二聚體與Pit-1反應元件的結合可增加GH-1基因的轉錄,從而促進GH的合成和分泌,Pit-1對於維持垂體GH細胞的增殖也具有重要的作用,因此,Pit-1基因的突變可引起GH細胞的萎縮及GH的缺乏,Pit-1蛋白以二聚體形式發揮作用,這是其突變以顯性方式遺傳的基礎,如果來自雙親一方的Pit-1基因發生突變,表達的蛋白質失去功能,這種無功能的突變Pit-1蛋白與正常Pit-1蛋白形成的二聚體失去激活GH-1基因轉錄的功能,從而幹擾正常Pit-1蛋白的功能.這也是一種顯性負現象,因此,病人隻要有來自雙親任一方的Pit-1基因發生突變,就可發病,故表現為顯性遺傳,Pit-1在PRL及TSH基因的表達中也具有重要的作用,因此,Pit-1基因的突變也引起PRL和TSH缺乏,病人還可有LH,FSH甚至ACTH的缺乏.
轉錄因子Prop-1為Pit-1的表達所必需,Prop-1的突變可引起為Pit-1表達下降,因而也能引起GH及其他垂體激素的缺乏,轉錄因子Lhx-3(或稱P-LIM)和HesX1(或稱Rpx)的突變可影響垂體的發育,從而引起GH等垂體激素的缺乏.
某些先天發育畸形如無腦畸形,前腦無裂畸形,垂體缺如,神經垂體錯位,面中線發育缺陷,蛛網膜囊腫等也可影響下丘腦-垂體的功能,從而產生GH缺乏癥.
下丘腦和垂體的腫瘤,炎癥,刨傷,手術,放射等均可導致GH不足,這些可統稱為獲得性GH缺乏癥.

生長激素缺乏癥 E23.013 GHD
生長激素缺乏癥 E23.013 GHD症状
生長激素缺乏癥的癥狀
閉經骨骺提前閉合聲調似小孩糖類皮質激素分泌過少毛發色淡而呈棕色無胡須抑鬱隱睪
原發性生長激素缺乏癥多見於男孩,患兒出生時身高體重正常,數周後出現生長發育遲緩,2-3歲後逐漸明顯,其外觀明顯小於實際年齡,但身體各部位比例尚勻稱,智能發育亦正常.身高低於同年齡正常兒童30%以上.隨年齡增長可出現第二性征缺乏和性器官發育不良.繼發性生長激素缺乏癥可發生於任何年齡,除有上述癥狀外,尚有原發病的種種癥狀和體征.
特發性生長激素缺乏癥多見於男孩,男:女=3:1,患兒出生時升高和體重均正常,1歲以後出現生長速度減慢,生長落後比體重低更為嚴重,升高低於同年齡、從性別正常健康兒童生長曲線第三百分位數以下(或低於兩個標準差),身高年增長速度小於4cm,智能發育正常.患兒頭顱圓形,面容幼稚,臉圓胖、皮膚細膩,頭發纖細,下頜和頦部發育不良,牙齒萌出延遲且排列不整齊,患兒雖生長落後,但身體各部比例勻稱,與其實際年齡相符.多數青春期發育延遲.
一部分生長激素缺乏患兒同時伴有一種或多種其他垂體激素缺乏,這類患兒除生長遲緩外,尚有其他伴隨癥狀:伴有促腎上腺皮質激素(ACTH)缺乏者容易發生低血糖,伴促甲狀腺激素(TSH)缺乏者可有食欲不振、不愛活動等輕度甲狀腺功能不足的癥狀,伴有促性腺激素缺乏者性腺發育不全,出現小陰莖(即拉直的陰莖長度小於2.5cm),到青春期仍無性器官和第二性征發育等.
器質性生長激素缺乏癥可發生於任何年齡,其中由圍生期異常情況導致者,常伴有尿崩癥狀.值得警惕的是顱內腫瘤則多有頭痛、嘔吐、視野缺損等顱內壓增高和視神經受壓迫的癥狀和體征.

生長激素缺乏癥 E23.013 GHD
生長激素缺乏癥 E23.013 GHD检查
生長激素缺乏癥檢查項目
染色體精氨酸胰島素生長激素放射免疫分析血糖血壓
實驗室檢查:
1、生長激素刺激實驗
生長激素缺乏癥的診斷依靠GH測定,正常人血清GH值很低,且呈脈沖式分泌,受各種因素影響,故隨意取血測血GH對診斷沒有意義,但若任意血GH水平明顯高於正常(>10μg/L),可排除GHD.因此,懷疑GHD兒童必須作出GH刺激試驗,以判斷垂體分泌GH的功能.
生理試驗系篩查試驗、藥物試驗為確診試驗.一般認為在試驗過程中,GH的峰值<10μg/L,即為分泌功能不正常.GH峰值<5μg/L,為GH無完全缺乏.GH峰值5-10μg/L,為GH部分缺乏.由於各種GH刺激試驗均存在一定局限性,必須兩種以上藥物刺激試驗結果都不正常時,才可確診為GHD.一般多選擇胰島素加可樂定或左旋多巴試驗.對於年齡較小的兒童,尤其空腹時有低血糖癥狀者給胰島素要特別小心,因其易引起低血糖驚厥等嚴重反應.此外,若需區別病變部位是在下丘腦還是在垂體,須作GHRH刺激試驗.
2、血GH的24H分泌譜測定
正常人生長激素峰值與基值差別很大,24h的H分泌量才能比較正確反映體內GH分泌情況,尤其是對GHND患兒,其GH分泌能藥物刺激試驗可為正常,但其24h分泌量則不足,夜晚睡眠時的GH峰值亦低,但該方案煩瑣、抽血次數多,不意為病人接受.
3、胰核樣生長因子(IGF-1)的測定
-1主要以蛋白結合的形式(IGF-BPs)存在於血循環中,其中以IGF-BP3為主(95%以上),IGF-BP3有運送和調節IGF-1的共冷,其合成也受GH-IGF軸的調控,因此IGF-1和IGF-BP3都是檢測該軸功能的指標,兩者分泌模式與GH不同,呈非脈沖式分泌,故甚為穩定,其濃度在5歲以下小兒甚低,且隨年齡及發育表現較大,青春期達高峰,女童比男童早兩年達高峰,目前一般可作為5歲到青春發育期前兒童GHD篩查檢測,該指標有一定的局限性,還受營養狀態,性發育成都和甲狀腺功能狀況等因素的影響,判斷結果時應註意.
4、其他輔助檢查
線檢查:常用右手腕掌指骨片評定骨齡.GHD患兒骨齡落後於實際年齡2歲或2歲以上.
或MRI檢查:已確診為GHD的患兒童,根據需要選擇頭顱CT或MRI檢查,以瞭解下丘腦-垂體有器質性病變,尤其對腫瘤有重要意義.
5、其它內分泌檢查
一旦確立,必須檢查下丘腦-垂體軸的其他功能,根據臨床表現可選擇測定TSH、T4或促甲狀腺素釋放激素(TRI)刺激試驗和促黃體生成速釋放激素(LHRH)此外試驗以判斷下丘腦-垂體、甲狀腺軸和性腺軸的功能.
主要診斷依據:①身材矮小,身高落後於同年齡、同性別正常兒童第三百分位數以下,②生長緩慢,生長速度<4cm/年,③骨齡落後於實際年齡2年以上,④GH刺激試驗示GH部分或完全缺乏,⑤智能正常,與年齡相稱,⑥排除其他疾病影響.
生長激素缺乏癥 E23.013 GHD预防
缺乏癥有明顯的傢族遺傳特點,可以做染色體檢查.
定期做好圍產期保健,避免圍生期病變史如:難產,宮內窒息等,以免造成腦部受損.
生長激素缺乏癥 E23.013 GHD治疗
一)治療
1、生長激素
基因重組人生長激素(rhGH)替代治療已經被廣泛應用,目前大都采用0.1U/kg,每日臨睡前皮下註射一次,每周6-7次的方案.治療應持骨骺愈合為止.治療時年齡越小,效果越好,以第一年效果最好,年增長可達10cm以上,以後生長速度逐漸下降,在用rhGH治療過程中可出現甲狀腺速缺乏,故須監測甲狀腺功能,若有缺乏適當加用甲狀腺素同時治療.
應用rhGH治療副作用較少,主要有:①註射局部紅腫、與rhGH物質基純度不夠以及個體反應有關,停藥後可消失,②少數註射後數月會產生抗體,但對促生長療效無顯著影響,③較少見的副作用有暫時性視乳頭水腫、顱內高壓等,④此外研究發現有增加股骨頭骺部滑出和壞死的發生率,但危險性相當低.
惡性腫瘤或有潛在腫瘤惡變者,嚴重糖尿病患者禁用rhGH.
2、促生長激素釋放激素
目前已知很多GH缺乏屬下丘腦性,故應用GHRH可奏效對GHND有較好療效,但對垂體性GH缺乏者無效.一般每天用量8~30μg/kg,每天分早晚1次皮下註射或24h皮下微泵連續註射.
3、口服性激素
蛋白同化類固醇激素有:①氟羥甲睪酮,每天2.5mg/m2;②氧甲氫龍,每天01-0.25mg/kg,③吡唑甲氫龍,每日0.05mg/mg,均為雄激素的衍生物,其合成代謝作用強,雄激素的作用弱,有加速骨骼成熟和發生男性化的副作用,故應嚴密觀察骨骼的發育,苯丙酸諾龍目前已較少應用.
同時伴有性腺軸功能障礙的GHD患兒骨齡達12歲時可開始用性激素治療,男性可註射長效庚酸睪酮25mg,每月一次,每3月增加25mg,直至每月100mg;女性可用炔雌醇1~2μg/日,或妊馬雌酮自每日0.3mg起?酌情逐漸增加,同時需監測骨齡.
二)預後
若該病不能及時診斷和治療,將會導致成年後身材顯著矮小、心血管疾病發生率升高,而且有相當多病例伴有性腺發育不良、中樞性甲狀腺功能低下、促腎上腺皮質激素(ACTH)缺乏癥.因此,不能得到醫治的生長激素缺乏性侏儒癥(GHD)將會嚴重地影響今後的工作、學習、婚姻、心理和生活質量等,如能得到早期治療,可以將身高達到正常人高度范圍內.另外,對維持肌肉活力、改善心臟功能、延緩衰老、防治骨質疏松、治療肥胖等也起著重要作用.
下丘腦-垂體部位腫瘤引起者,可出現視力減退,視野缺損,後期可出現顱內壓力增高的表現,以及嗜唾、抽搐.
生長激素缺乏癥 E23.013 GHD饮食
體育鍛煉可加強機體新陳代謝過程,加速血液循環,促進生長激素分泌,加快骨組織生長,從而有益於人體長高.鼓勵孩子蹦蹦跳跳是促進長身體的積極因素,能使身高的生理效應得到最大限度發揮.飲食不當晚飯吃得過多,吃的食物不易消化,孩子胃裡不舒服,睡眠就會受到影響.如果晚飯吃得過少,孩子就可能因饑餓而不能入睡,也可能渴瞭想喝水.
生長激素缺乏癥 E23.013 GHD并发症
繼發性生長激素缺乏癥除上述表現外,可伴有原發病的各種癥狀,由下丘腦-垂體部位腫瘤引起者,可出現視力減退,視野缺損,後期可出現顱內壓增高的表現,以及嗜睡,抽搐等.
1/2 1 2 下一页 尾页


















