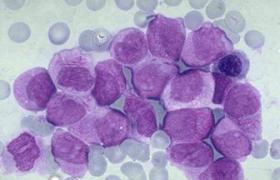
慢性粒細胞白血病
慢性粒細胞白血病百科
慢性髓性白血病(CML)是骨髓造血幹細胞克隆性增殖形成的惡性腫瘤,絕大多數患者緩慢起病,早期常無癥狀,臨床逐步出現乏力、食欲不振、腹部脹滿、盜汗和體重降低,偶因體檢發現白細胞計數增高或左上腹包塊而作進一步檢查.通常大多數CML患者臨床上處於“慢性”或“穩定”階段,此期可持續3~4年.常見的癥狀包括:貧血、脾區不適、出血及乏力、體重減輕和低熱等代謝增高的表現.部分患者無癥狀,因常規體檢發現白細胞數、血小板數增高或脾臟腫大而診斷.少數病人有痛風性小關節疼痛.此外,還有視力障礙、神經系統病變以及陰莖異常勃起等.慢性期患者不易感染,發熱少見.當疾病進展時,患者開始出現發熱、骨痛、脾大等現象,白細胞計數持續上升,骨髓或外周血中原始細胞增多等表現.臨床上可分為慢性期和加速期、急變期.
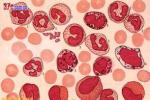
慢性粒細胞白血病
慢性粒細胞白血病病因
一、發病原因
1、離子輻射可以使CML發生率增高,在廣島和長崎原子彈爆炸後幸存者、接受脊椎放療的強直性脊椎炎患者和接受放療的宮頸癌患者中CML發病率與其他人群相比明顯增高.
2、長期接觸苯和接受化療的各種腫瘤患者可導致CML發生,提示某些化學物質亦與CML發關.
3、CML患者HLA抗原CW3和CW4頻率增高,表明其可能是CML的易感基因.
4、盡管有傢族性CML的報道,但CML傢族性聚集非常罕見,此外單合子雙胞胎的其他成員CML發病無增高趨勢,CML患者的父母及子女均無CML特征性Ph染色體,說明CML是一種獲得性白血病.
二、發病機制
1.起源於造血幹細胞CML是一種起源於造血幹細胞的獲得性克隆性疾病,其主要證據有:
①CML慢性期可有紅細胞,中性粒細胞,嗜酸/嗜堿粒細胞,單核細胞和血小板增多;
②CML患者的紅系細胞,中性粒細胞,嗜酸/嗜堿粒細胞,巨噬細胞和巨核細胞均有Ph染色體;
③在G-6-PD雜合子女性CML患者中,紅細胞,中性粒細胞,嗜酸/嗜堿粒細胞,單核細胞和血小板表達同一種G-6-PD同工酶,而成纖維細胞或其他體細胞則可檢測到兩種G-6-PD同工酶;
④每個被分析的細胞其9或22號染色體結構異常都一致;
⑤分子生物學研究22號染色體斷裂點變異僅存在於不同CML患者,而在同一病人的不同細胞中其斷裂點是一致的;
⑥應用X-連鎖基因位點多態性及滅活式樣分析亦證實瞭CML為單克隆造血.
2.祖細胞功能異常相對成熟的髓系祖細胞存在有明顯的細胞動力學異常,裂指數低,處於DNA合成期的細胞少,細胞周期延長,核漿發育不平衡,成熟粒細胞半衰期比正常粒細胞延長,采用3H自殺試驗證實僅隻有20%的CML集落處於DNA合成期,而正常人為40%,CML原粒,早幼粒細胞標記指數比正常人低,而中,晚幼粒細胞標記指數與正常對照相比無明顯差別,造血祖細胞集落培養發現CML骨髓祖細胞與外周血祖細胞的增殖能力不同,骨髓CFU-GM和BFU-E數與正常對照相比通常增高,但亦可正常或減低,而外周血可升高至正常對照的100倍,Ph陽性CML病人骨髓細胞長期培養研究發現,經幾周培養後在培養基中可檢測到Ph陰性的祖細胞,現已證實這主要為CML造血祖細胞黏附功能異常所致.
3.分子病理學1960年,Nowell和Hungerfor描述瞭CML相關的Ph染色體,這是首次發現的與一特異人類腫瘤相關的非隨機染色體異常,1973年Rowley采用奎寧和姬姆薩染色技術首次證實CML中發現的Ph染色體(22q-異常)是t(9;22)(q34;q11)染色體易位所致,1982年在9q34斷裂區克隆出瞭ABL基因,1983年證實位於q34的基因片段易位到22號染色體上與22q11斷裂區一個稱為BCR的基因形成BCR-ABL融合基因.
(1)ABL基因:原癌基因c-abl定位於q34,在物種發育過程中高度保守,編碼在所有哺乳動物組織和各種類型細胞中均普遍表達的一個蛋白質,c-abl長約230kb,含有11個外顯子,走向為5′端至著絲粒,該基因第一個外顯子有兩種形式,外顯子1a和1b,因而有兩種不同的c-ablmRNA,第一種稱為1a-11,長6kb,包括外顯子1a-11;另一種稱為1b,自外顯子1b開始,跨越外顯子1a和第一個內含子,同外顯子2-11相接,長為6kb,這兩種ABL的RNA轉錄編碼兩種不同的分子量均為145000的ABL蛋白,DNA序列分析發現,c-abl屬非受體蛋白-酪氨酸激酶傢族,除激酶片斷外,該基因還有在信號傳導蛋白的相互作用和調節中非常重要的SH2和SH3片斷,c-abl的特征是有一個大的C末端非催化片斷,該片斷含有DNA和細胞骨架結合的重要序列和一個參與該傳導信號的區域,正常的p145ABL穿梭於細胞核和胞漿之間,主要定位於細胞核,具有較低的酪氨酸激酶活性,p145ABL的活性和細胞內定位受連接細胞骨架與細胞外間質的整合素(integrins)調控,現有研究表明至少在纖維細胞,ABL激活需要細胞黏附,因此ABL可能通過將整合素信號傳遞至細胞核從而充當黏附和細胞周期信號之間的橋梁,參與細胞生長和分化控制.
(2)BCR基因:BCR基因定位於22q11,長130kb,有21個外顯子,起始方向5′端至中心粒,有4.5kb和6.7kb兩種不同的BCRmRNA轉錄方式,編碼一分子量為160000的蛋白p160BCR,該蛋白有激酶活性,p160BCR之C末端與ras相關的GTP結合蛋白p21的GTP活性有關聯.
(3)BCR-ABL基因:位於9q34的c-abl基因易位於22號染色體與位於22q11的bcr基因形成BCR-ABL融合基因,迄今CML患者中已發現有3個bcr斷裂點叢集區,分別為M-bcr,m-bcr,u-bcl和6種BCR-ABL融合轉錄方式,與M-bcr相應的有b2a2,b3a2,b2a3,其編碼蛋白為p210,與m-bcr相應的有ela2,其編碼蛋白為p190,與u-bcr相應的有e19a2,其編碼蛋白為p230.
小鼠模型體內已證實BCR-ABL可導致CML發生,BCR-ABL融合蛋白定位於細胞漿,具有極高的酪氨酸激酶活性,通過改變作為BCR-ABL催化底物的一些關鍵的調節蛋白磷酸化狀況激活多種信號傳導途徑,如通過激活參與細胞增殖和分化調控的Ras信號途徑,使祖細胞數量增多,幹細胞池減少,幹細胞成為增殖池的一部分,從而使未成熟粒細胞不斷擴增,BCR-ABL作用的另一種機制是改變正常整合素功能,正常造血祖細胞黏附於細胞外基質,而黏附是由祖細胞細胞表面受體特別是整合素來介導的,BCR-ABL通過幹擾β1整合素的功能導致CML細胞的細胞黏附功能缺陷,從而使未成熟細胞釋放至外周血並遷移至髓外部位.
最近,CML發病機制研究又取得瞭進展:
①體外培養發現,BCR-ABL通過抑制凋亡而延長CML祖細胞的因子非依賴性生長時間;
②用反義寡核苷酸下調BCR-ABL表達後可能通過增加細胞對凋亡的敏感性從而抑制白血病細胞在小鼠體內生長,特別是減少CML病人早期祖細胞集落形成,降低CML樣細胞系的細胞增殖;
③表達BCR-ABL的,轉化的,因子非依賴性的,可致瘤的小鼠造血細胞通過上調bcl-2而增加對凋亡的敏感性,當bcl-2表達受抑後,BCR-ABL陽性細胞又變成瞭因子依賴性和非致瘤性,以上實驗結果表明,BCR-ABL抑制細胞凋亡導致髓系細胞不斷擴增是CML的又一發病機制.
(4)急變發生機制:細胞遺傳學研究發現80%的AP或BPCML患者有繼發性染色體異常,最常見的異常依次為+8,+Ph,i(17),+19,+21和-Y,急性粒細胞白血病變(急粒變)的患者中約80%有非隨機性染色體異常,其染色體核型常表現為超二倍體,最常見的異常為+8,且+8常與其他染色體異常如i(17),+Ph,+19等同時出現,其次為+Ph,i(17)和-Y,急性淋巴細胞白血病變(急淋變)的患者約30%有繼發性克隆性染色體異常,常為染色體丟失,從而表現為亞二倍體或結構異常,常見異常為+Ph和-Y,+8少見,i(17)尚未見報道,-7,14q+與急淋變特異相關,盡管有研究發現急變期CML有N-Ras基因突變和c-Myc基因表達增高,但其發生率極低,Rb基因在急變期CML患者亦極少有改變,Sill等發現p161NK4A基因的純合子缺失與CML急淋變相關,CML急性變分子機制研究較多的還是p53基因,20%~30%的急粒變的患者存在有p53基因結構和表達的異常,CMLp53基因改變特征為:①主要改變是基因重排和突變;②主要見於急粒變,急淋變極少見;③p53突變常見於有17P-異常患者;④p53突變能導致CML的急粒變,最近,又有鈣調素基因甲基化程度,端粒長度和端粒酶活性改變與CML急變關系的研究報道,但其意義尚待進一步闡明.
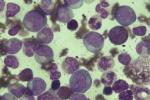
慢性粒細胞白血病
慢性粒細胞白血病症状
1.慢性期
(1)癥狀:通常大多數CML患者臨床上處於“慢性”或“穩定”階段,此期可持續3~4年,常見的癥狀包括:貧血,脾區不適,出血及乏力,體重減輕和低熱等代謝增高的表現,20%~40%的患者無癥狀,因常規體檢發現白細胞數,血小板數增高或脾臟腫大而診斷,少數病人有痛風性小關節疼痛,此外,還有視力障礙,神經系統病變以及陰莖異常勃起等,慢性期患者不易感染,發熱少見.
(2)體征:主要表現為臟器浸潤,90%患者脾臟腫大,程度不一,肋下可及至巨脾伸延至盆腔,質硬常有明顯切跡,脾栓塞時脾區可觸及摩擦感或聞及摩擦音,可有輕到中度肝臟腫大,淋巴結腫大少見,胸骨常有壓痛,以胸骨柄的下端為著,眼底視網膜浸潤,可見到視網膜血管迂回擴張,並可見呈片狀的出血斑以及白色浸潤中心.
2.急變期
慢性期經過數月或數年之後,惡性造血幹細胞極度增生,骨髓原粒+早幼粒細胞≥20%,可伴由血小板衍生生長因子過多引起的骨髓纖維化改變,每個病人何時急變尚不能預測,一旦發生急變,病情迅速惡化,治療非常困難,存活期很少超過6~12個月.
(1)癥狀:有不明原因的發熱,脾進一步腫大;出現骨痛,出血以及髓外腫物等浸潤現象,如淋巴結腫大,皮膚軟組織腫塊或溶骨性病變.
(2)急變類型:
①約65%為急粒變:包括:A.原始粒細胞危象,病情突然驟變,骨髓或血液中出現大量的原始粒細胞,原粒+早幼粒>90%,病情發展快,病程短,一般在1~2個月內死亡;B.慢粒急變,指CML經數周至數月的轉變過程,出現急性白血病的所有征象,骨髓中原始+早幼粒>20%,對治療耐藥,生存期不超過6個月.
②約30%為急淋變:包括普通型急性淋巴細胞白血病(C-ALL),非T非B淋巴細胞白血病,前B細胞白血病及B,T細胞白血病,急淋變經過長春新堿及潑尼松可獲暫時的緩解,但最終在0.5~1年內死亡.
③5%為其他少見類型的急髓變:包括組織細胞變,紅白血病變,巨核細胞變及急性單核細胞變,血象,骨髓象,細胞形態學等改變有其相應的特征,且預後差,絕大多數患者急變後6個月內死亡.
3.加速期
介於慢性期和急性期之間,此期臨床開始出現低熱,脾大等現象,貧血逐漸加重,白細胞持續上升,幼稚細胞開始增多,原粒+早幼粒≥10%,對原來有效的藥物出現耐藥,在數周或數月內即可演變成典型的急性期,染色體在此期已有變化如急性期,故染色體的改變早於血液學和臨床的轉變,可作為疾病進展及預後判斷的指標.
典型的CML伴有脾大,外周血白細胞數增高,可見各階段幼稚粒細胞,嗜酸和嗜堿粒細胞增高,骨髓增生明顯或極度活躍,以粒細胞系增生為主,中性晚幼及桿狀核粒細胞明顯增生,嗜酸和(或)嗜堿粒細胞亦增多,巨核細胞系常增生,中性粒細胞堿性磷酸酶積分(ALP)減低,細胞遺傳學檢查有Ph染色體或應用分子生物學方法檢測出BCR-ABL基因重排或融合,診斷並不困難.

慢性粒細胞白血病
慢性粒細胞白血病检查
1.慢性期
(1)血象:白細胞數常>50×109/L有時可達500×109/L以上,約1/3患者血紅蛋白<110g/L,貧血多為正常細胞正色素性,血小板往往增多,有時高達1000×109/L,少數病人可正常減少,血塗片檢查中可見不同成熟階段的粒細胞,以中,晚幼粒細胞階段居多,原料細胞<5%,原粒+早幼粒細胞≤10%,嗜酸性及嗜堿性粒細胞增多,有少量有核紅細胞出現.
(2)骨髓象:增生極度活躍或明顯活躍,以粒系為著,粒與紅之比可增至10∶1~20∶1,粒系各階段均增加,以中,晚幼粒細胞增加為主,嗜酸性與嗜堿性粒細胞比例明顯高於正常,巨核細胞及血小板亦增多.
(3)中性粒胞堿性磷酸酶(ALP):染色積分減低或接近於零.
(4)細胞遺傳及分子生物學檢查:90%以上的慢性期患者骨髓中期分裂細胞往往Ph染色體陽性,分帶技術證明9號染色體長臂3區4帶與22號染色體1區1帶部分片段相互易位,即t(9;22)(q34;11),熒光素染色體原位雜交術(FISH)敏感性更高,提取骨髓或外周血單個核細胞的DNA,經DNA印跡法可檢測到bcr基因重排,發生在5端(b3a2),若提取骨髓或血單個核細胞部RNA,經反轉錄聚合酶鏈反應(RT-PCR)術可檢測到bcr/abl轉錄產物mRNA,是目前最靈敏而又特異的方法.
(5)血清生化測定:血清尿酸,乳酸脫氫酶及溶菌酶往往增高.
2.急變期
貧血迅速加重,骨髓及外周血中原始粒細胞明顯增多,骨髓原始粒細胞≥20%,如為急性危象則可達卡90%以上,血小板減少,中性分葉核細胞堿性磷酸酶可升高或正常,遺傳學檢查,常為非整倍體,除t(9;22)(q341;q11)的Ph染色體外,還附加其他染色體的異常,如出現第二個Ph染色體,或多一個8號染色體(+8),或者說17號染色體長臂等臂缺失(ISO17q-).
3.加速期
白細胞持續上升,幼稚細胞開始增多,原粒+早幼粒≥10%.
1.骨髓活檢病理切片銀染常示網狀纖維增生,約一半患者為顯著增生.
2.根據病情、癥狀、體征選擇做X線,CT,MRI,B超,心電圖等檢查.
慢性粒細胞白血病预防
避免或減少有害物質如放射性物質,化學物質,化學藥物的接觸.
慢性粒細胞白血病治疗
一、治療
CML的療效判斷包括血液學緩解、細胞遺傳學緩解(即Ph細胞消失率)和分子生物學緩解(即BCR-ABL融合基因轉陰率),由於此三種不同的緩解程度與CML患者的生存期顯著相關,因此現代CML治療的主要目的是如何提高後兩者的緩解率,爭取患者獲得長期無病生存.
1.常規治療
CML就診或復發時常有高尿酸癥,因此,治療前應予別嘌醇300mg/d,口服,並充分補液以維持尿量;如果患者有大量細胞溶解的危險因素,則別嘌醇給藥量及給藥次數均應增加,並應維持尿量在150ml/h.由於別嘌醇可出現過敏性皮炎,因此在白細胞數下降至正常、脾大明顯縮小、無明顯高尿酸血癥後應停用.
2.單藥化療
(1)白消安(馬利蘭):是第一個廣泛應用於CML治療的化療藥物.其療效於1968年經隨機比較得以肯定.常用劑量為4~6mg/d,口服.由於該藥有明顯的後效應,因此當白細胞計數下降至30×109/L左右應減量或停藥.大部分患者需維持治療,維持劑量可降至2mg,口服,2次/周,約95%的慢性期患者有效,白細胞計數下降、脾縮小、血細胞比容升高、一般狀況恢復正常.
白消安(馬利蘭)治療常不能使Ph染色體消失,白消安(馬利蘭)治療的目的是控制慢性期,減少死亡率.
該藥的主要副作用有嚴重骨髓抑制、皮膚色素沉著、乏力、發熱和腹瀉為特征的類似於腎上腺皮質功能不全的綜合征和肺纖維化.
(2)羥基脲:1993年通過隨機對照系列比較證實羥基脲(HU)優於白消安(馬利蘭).其中位生存期刪組明顯好於BUS組(分別為58個月和45個月),5年生存率分別為44%和32%.由於該藥毒性小,可延長CML慢性期和有利於患者進行造血幹細胞移植等優點,現已成為CML的首選化學治療藥物.依白細胞計數,起始劑量為1~4g/d,口服;當白細胞下降至20×109/L時改為l~2g/d,維持量為0.5~2.0g/d;當白細胞計數下降至5×109/L時應暫停.HU的副作用輕,可有皮膚丘疹、骨髓細胞巨幼變、大紅細胞增多、月經量增多、禿發等,但骨髓抑制少,沒有發生肺纖維化者.部分患者可有Ph染色體陽性率減低.最近有研究發現羥基脲(HU)可延緩CML患者骨髓纖維化的發生,對早期輕度纖維化患者有逆轉作用.
(3)靛玉紅及其衍生物甲異靛:靛玉紅和甲異靛是中國醫學科學院血液學研究所經過20多年研究首創用於治療CNL的新藥.單用靛玉紅100~300mg/d,分3~4次口服,總有效率為95.8%.單用甲異靛75~150mg/d,分3次口服,總緩解率為80.6%.與BUS和HU相比,其縮脾效果明顯好於前者.最近,研究證實甲異靛長期療效與HU相似,甲異靛聯合羥基脲(HU)可明顯延長患者慢性期,降低患者5年急變率.部分患者可有Ph染色體陽性率減低.主要副作用有不同程度的骨關節疼痛、惡心、納差、腹痛、腹瀉等消化道反應,極少在治療期間出現骨髓抑制.
(4)其他:國內外有單用環磷酰胺、巰嘌呤(6-巰基嘌呤)、氧芬胂(馬法蘭)、苯丁酸氮芥(瘤可寧)、二溴甘露醇、烏拉非汀(合520)、秋水仙胺、二溴衛矛醇、卡波醌、三尖杉堿等治療CML慢性期患者,這些藥物雖均對CML有效,但沒有一種藥物的療效比BUS或HU好.最近有長療程高三杉尖酯堿2.5mg/(m2·d),靜滴,第1~14天,使6%CML患者獲得完全細胞遺傳學緩解的報道.
3.幹擾素治療
1983年Talpaz等報道單用天然幹擾素(IFN)治療CMLCP患者51例,其中71%(36例)獲血液學緩解,且7例(14%)Ph染色體消失.此後關於天然和重組幹擾素治療CML的療效觀察表明血液學緩解率為61%~80%(中位64%),29%~65%的患者有不同程度的細胞遺傳學緩解,現已成為CML的首選治療藥物.
盡管迄今關於幹擾素(IFN)治療CML取得瞭一些共識:
①天然幹擾素與重組人幹擾素治療CML療效相似;
②持續用藥比間歇用藥好,大劑量比小劑量療效好,初治病例的血液學完全緩解明顯比復治者高,加速期的療效比慢性期差;
③肌內註射或皮下註射比靜脈註射好.但仍存在諸多問題尚待解決:A.幹擾素(IFN)是否可以延長CML患者生存期:最近發表的幹擾素(IFN)治療CML的大系列隨機對照研究結果不一致,意大利協作組和英國MCR的結果顯示幹擾素(IFN)治療組較羥基脲(HU)(或白消安(BUS))治療組生存期明顯延長,二者有顯著性差異,而德國CML研究組的結果則發現二者生存期並無區別(表2);B.幹擾素(IFN)的最適劑量和用藥時間:至今尚無一致意見,但一般為幹擾素(IFN)的起始劑量應為5MU/(m2·d),2~3周後劑量增至9~12MU/d,或達到獲顯著血液學療效[即WBC計數(2~4)×109/L,血小板計數接近50×109/L]的最大耐受量及患者出現毒性癥狀需要減少劑量.可望獲細胞遺傳學緩解的最短時間為6個月,一般用至病情進展或出現不可耐受的藥物毒性;C.幹擾素(IFN)種類與療效的關系:現一般認為各種不同種類的幹擾素a臨床療效無差別,幹擾素γ療效不清,幹擾素α聯合幹擾素γ不能提高療效;D.幹擾素(IFN)聯合其他化療藥物如羥基脲、小劑量阿糖胞苷20mg/(m2·d)×10天已有Ⅱ期臨床觀察,表明療效優於單用IFN.
幹擾素(IFN)治療CML的早期常見副反應有發熱、畏寒、流感樣癥狀、頭痛等,持續約幾天至2個月;晚期可有持續乏力、食欲下降、體重下降,少數病例可有貧血、血小板減少、肝腎功能損害、禿發,有時有骨骼、肌肉疼痛及甲狀腺功能低下、憂鬱等,嚴重者可有心絞痛、註意力不集中、記憶力減退及昏睡等神經系統毒性表現.劑量減少時以上癥狀可減輕或消失,給予小劑量解熱鎮痛藥如對乙酰氨基酚(撲熱息痛)等可解除上述副作用.
4.伊馬替尼(ST1571、格列衛)
1998年6月伊馬替尼(STl57l)(亦稱CGP57148或Gleevec)開始Ⅰ期臨床試驗,共83例幹擾素治療失敗的慢性期CML患者,按25~1000mg/d共分14個劑量組接受瞭治療,結果證實獲最大臨床療效的最低劑量為300mg/d,54例接受300mg/d或以上劑量的患者中53例(98%)獲完全血液學緩解,31%的患者獲顯著細胞遺傳學緩解.受此結果鼓舞,58例慢粒急變或Ph急性白血病患者接受瞭治療,劑量為300~1000mg/d,55%(21/38例)的CML急粒變和70%(14/20例)的PhALL患者獲血液學療效.其後454例CPCML、181例APCML和229例CML急粒變患者進入瞭Ⅱ期臨床試驗,完全血液學緩解率分別為91%、69%和29%,顯著細胞遺傳學緩解率分別為55%、24%和16%.2001年5月10日該藥獲美國FDA批準上市,現正在全球范圍進行Ⅲ期臨床試驗.
伊馬替尼(ST1571)的作用機制是抑制BCR-ABL融合基因的酪氨酸激酶活性.迄今,伊馬替尼(STl571)的最適臨床使用劑量尚不十分明確,CPCML的推薦起始劑量為400mg/d,加速期(急變期)推薦起始劑量為600mg/d,無效患者可增量至800mg/d.由於伊馬替尼(STl571)的半衰期為14~16h,因此1次/d給藥即可.主要副作用有骨髓抑制、惡心、肌肉痙攣、骨骼疼痛、關節痛、皮疹、腹瀉、水腫、體液瀦留和肝功能受損等.
5.聯合化療
采用阿糖胞苷、蒽環類藥物、硫鳥嘌呤(6-TG)、環磷酰胺、門冬酰胺酶、巰嘌呤(6-MP)、依托泊苷(VP-16)和白消安(馬利蘭)等藥物組成不同的聯合強烈化療方案治療CML,Ph染色體減少率高於常規單藥治療.但絕大部分研究表明並不能明顯延長生存期(表3).最近人們又采用聯合化療加幹擾素a療法試圖提高生存率和細胞遺傳學緩解率,但迄今結果並不理想.
6.造血幹細胞移植(SCT)
(1)自體幹細胞移植(ASCT):CPCML患者采用ASCT的結果表明,CP期進行凈化幹細胞的ASCT能明顯延長CML的生存期.
近年來,人們采用聯合化療動員Ph-外周血幹細胞,幹擾素α單獨或聯合羥基脲治療CML等“體內凈化”和長期骨髓細胞培養、4-HC和ASTA-Z等藥物、幹擾素、反義寡核苷酸等“體外凈化”方法來篩選Ph-外周血幹細胞,盡管使ASCT後Ph轉陰率有所提高,但凈化幹細胞移植患者的生存率並無明顯提高.最近,McGlare等總結分析歐美8個BMT中心報告的ASCT治療CML的效果,200例患者中,CP142例,AP30例,BP或第二次慢性期((CP2)28例,中位年齡42歲,診斷至移植的中位時間為15個月,123例幹細胞來源於骨髓,73例來源於外周血,有21例骨髓經體外10天培養凈化,23例骨髓經幹擾素γ凈化,移植後中位隨訪48個月,CP期移植的患者5年生存率為95%±5%,AP期為27%±10%,而急變後移植的患者全部於移植後2年半內死亡.預後分析表明:年齡>40歲和診斷至移植的時間過長為不利因素,而幹細胞來源(骨髓或外周血)及“體外凈化”對生存無影響.
(2)異體幹細胞移植(Allo-SCT):迄今異體骨髓移植(Allo-BMT)是惟一可以治愈CML的手段.CP移植的存活率比在AP或BP好,且復發率低.CP、AP、BP移植後3年存活率分別為55%~70%、10%~30%、0~20%,復發率分別為20%、50%和75%.BMT前接受過BUS治療的患者療效差,且3年無病生存率(DFS)為45%,而接受過HU治療者則可為61%.IFN-a治療對BMT治療療效無影響.各種預處理方案如Cy+TBI、白消安(BUS)+Cy對療效無明顯影響.Allo-BMT的主要移植相關死亡原因是GVHD.去T細胞Allo-BMT雖然可以降低GVHD發生率,但復發率則明顯增高,提示移植物抗白血病(GVL)效應是Allo-BMT治療CML顯示療效的重要因素.
盡管Allo-BMT治療CML取得瞭滿意的效果,但僅有20%~25%的患者有HLA匹配的同胞供體.近年來,對無關供體的Allo-BMT治療CML的研究已取得瞭可喜的成績,2年DFS為31%,但其移植失敗率高達16%,Ⅲ和Ⅳ級急性GVHD發生率約為54%,嚴重慢性GVHD亦高達52%.
最近已有研究表明應用異體外周血幹細胞移植比Allo-BMT、移植後髓系和免疫重建要快,二者近期療效相似,遠期療效尚待確定.此外,相關的/無關的臍血移植、非骨髓清除性造血幹細胞移植也有初步報道.
為瞭更好地指導臨床選擇合適的患者進行異體外周血和骨髓移植,歐洲外周血和骨髓移植組根據3142例患者的資料,提出瞭預後判定積分系統(表4).根據該積分系統,積分為0、1、2、3、4、5和6的患者其5年無病生存率分別為72%、70%、62%、48%、40%、18%和22%,移植相關死亡率為20%、23%、31%、46%、51%、71%和73%.
異基因移植後復發患者的治療包括第2次移植,rIFN-α和供體白細胞輸註(donorLeukocyteinfusion,DLI可使約75%患者再獲CR,血液學復發患者8年存活率可達60%,細胞遺傳學復發患者8年存活率約80%,達細胞遺傳學和分子生物學療效的中位時間為4~6個月,那些細胞遺傳學/血液學復發者、移植時為第一次慢性期(CPI)、移植後緩解時間1年以上、移植後無慢性GVHD、為嵌合體造血的患者療效較佳.
7.急變期的治療
急性髓系細胞變患者可采用原發性AML治療方案,但CR率<20%,且完全緩解期僅幾周或幾個月.25%~35%的急變患者為急淋變或雙表型白血病變,盡管采用VDLP方案約60%的患者有效,約有1/3患者可返回CP2,但其總生存率亦僅為4~6個月,造血幹細胞移植3年DFS可達15%~20%.
8.新的治療方法
(1)“良性”祖細胞篩選和擴增:已有實驗證實在CML患者骨髓CD34、HLA-DR-組分中可以富集Ph-的造血祖細胞,因此可以據此篩選非白血病性幹/祖細胞,通過體外擴增後用於移植.另一種方法是根據“正常”細胞和白血病細胞對細胞因子的不同反應,用幹細胞因子缺陷的基質來進行長期培養有利於“良性”細胞增長等特征來進行細胞的功能篩選.可以預測,隨著富集和擴增方法的不斷改進,富集Ph-細胞進行體外擴增後用於移植是今後CML的治療方向之一.
(2)反義寡核苷酸:以BCR/ABL為靶標設計的反義寡核苷酸(amtisenseoligonucleotide)可以降低BCR/ABL的轉錄水平和體外培養的CML細胞的增長(可能通過誘導凋亡),現主要用作CML自身幹細胞移植的“凈化”.已有用BCR/ABL和C-MYB反義寡核苷酸體外凈化後骨髓成功植活和獲部分細胞遺傳學緩解的初步報道.反義寡核苷酸聯合化療藥物方案現已在SCID小鼠動物實驗證實可顯著延緩白血病的發生.
(3)基因治療:已有用反轉錄病毒載體構建的BCR/ABL反義基因聯合一個甲氨蝶呤(MT)X耐藥基因的所謂“雙基因治療策略”的報道,體外實驗結果表明該方法可用於CML自身幹細胞移植體外凈化和移植後化療,以進一步根除微小殘留病.
(4)免疫調節治療:現已有具有免疫源性的P210BCR/ABL融合片段和結合主要組織相容性工類抗原等位基因復合物多肽的報道,亦已建立識別BCR/ABL表達細胞的肽特異性CD4T細胞系,體外實驗證實利用肽特異性CD4T細胞可以使P210b3a2產物降解.這些結果提示可以用人T細胞介導的腫瘤相關抗原的識別來進行CML的治療.此外,有治療潛能的還有白介素-2激活NK細胞和細胞毒T細胞.CML患者自身NK細胞能抑制CML祖細胞生長,因此,可利用自身激活的NK細胞經體外擴增後用於自身幹細胞移植凈化和CML免疫治療.最近,又有實驗發現CML患者骨髓體外培養獲得的樹狀突細胞能刺激自身細胞,並具有抗增殖作用,而抗正常骨髓活性極低,提示該方法可用於CML的過繼免疫治療.
9.治療策略的選擇
隨著CML治療方法的不斷增多,如何根據病人具體情況制訂出一個最佳的個體化治療方案已成為每位血液學工作者面臨的一個問題,已提出瞭幾個CML治療選擇模式;最近美國血液學會組織全世界CML專傢詳盡分析瞭迄今發表的CML治療較有價值的文獻資料,提出一個CML治療指南:
①首先根據患者年齡、身體狀況,是否有相關的或無關的骨髓供體決定是否進行幹細胞移植;
②如果選擇非移植治療方案則應作出一個詳盡的給藥方案,如,用rIFN-a,必須對其劑量、療程是否聯用羥基脲或阿糖胞苷作出決定;
③應制定一個系統計劃來觀察細胞遺傳學療效程度和起效時間及分子生物學療效;
④一旦明確瞭診斷和治療意見,作出涉及治療療效和病人意願的改動時必須經過審查.
二、預後
目前尚無可靠的預測方法,但是大量研究證實:年齡、脾臟大小、血小板計數、外周血或骨髓中原始細胞比例、嗜堿性粒細胞比例均為影響預後的因素.1984年Sokal提出一個預測危險比率(riskratio,RR)的公式,以年齡(年)、脾(肋中線下cm)、血小板數(109/L)及血中原粒細胞數(%)為變量.公式如下:
RR={0.0116(年齡-43.4)+0.0345(脾-7.51)+0.188[(血小板數/700)2-O.563]+0.087(原粒-2.10)}
RR<0.8為低危,RR<0.8~1.2為中危,RR>1.2為高危.有資料表明低危組中位存活期為60個月,中危組為46個月,高危組32個月.但是對於接受IFN治療的患者,此公式的判斷結果不肯定,而對IFN治療的反應則是影響預後的重要因素.
慢性粒細胞白血病饮食
1、多以清淡食物為主,註意飲食規律.
2、根據醫生的建議合理飲食.
3、該疾病對飲食並沒有太大的禁忌,合理飲食即可.
慢性粒細胞白血病并发症
1、慢性期部分病人可出現脾栓塞、脾破裂、脾出血.
2、加速期、急變期可合並感染,發熱,貧血性心臟病,心衰等並發癥,急性痛風性關節炎可並發肺,中樞神經系統,某些特殊感覺器官和陰莖等循環血管內血流受阻,出現相應的癥狀和體征,如呼吸急促,呼吸困難,發紺,頭暈,語言不清,譫妄,昏迷,視物模糊,耳鳴,聽力減退及陰莖異常勃起.
1/2 1 2 下一页 尾页


















