小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血
小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血百科
β地中海貧血(β-mediterraneananemia)是指β鏈的合成受部分或完全抑制的一組血紅蛋白病.

小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血
小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血病因
1.病因
β珠蛋白基因位於11號染色體短臂1區2帶(簡記:11P12),本病除少數幾種為幾個核苷酸缺失外,絕大部分都是點突變(單個核苷酸置換,增加或缺失)所致,全世界已發現100種基因突變類型,我國有20種(表1),突變致β鏈合成部分受抑制者稱“β地中海貧血";致β鏈完全受抑制者稱“βo地中海貧血",肽鏈合成的抑制涉及δ鏈,稱δβ海洋性貧血(δβ或δβo),染色體上的2個等位基因突變點相同者稱純合子;等位基因的突變不同者稱“雙重雜合子";同源的染色體上隻有1個突變者稱“雜合子".
2.分類
根據β基因缺陷所產生的雜合子和純合子的不同,其臨床表現亦有差異,按照病情輕重的不同,可將β地中海貧血為重型,輕型和中間型三種類型.
(1)重型β地中海貧血:重癥β地中海貧血β-mediterraneananemiamajor)又稱庫理貧血(Cooleysanemia),這是β地中海貧血純合子,β和β地中海貧血雙重雜合子或β地中海貧血純合子或δβo純合子,是我國常見的一種地中海貧血,本癥患者雙親均為β-地貧雜合子,其子女中獲得重癥β-地貧幾率為25%,50%為雜合子,餘25%為正常.
(2)中間型β地中海貧血:中間型β海洋性貧血(β-mediterraneananemiaintermedia)包括:β基因純合子,某些β0/β雙重雜合子,非典型β地貧雜合子,合並α地貧血和δβ地貧,其臨床表現介於重型與輕型之間,特點:
①發病年齡較晚(常於4~5歲).
②中度貧血,Hb60~90g/L.
③輕度肝脾腫大.
④外周血塗片紅細胞形態與重型類似.
⑤HbF含量增高,HbA2可稍高,正常或降低.
⑥輸血量小或不必輸血仍可維持生命;傢系調查證實為上述基因傳遞.
非典型β地貧雜合子:指位於β珠蛋白基因遠側的啟動區域位點控制區(locuscontrolregion,LCR),這些並非在β鏈上的分子缺陷(非典型β地貧基因)與典型的β地貧基因組合形成雙重雜合子.
(3)輕型β-地中海貧血:輕型β-地中海貧血(β-mediterraneananemiaminor或trait)是β0,β或δβ基因,包括雙重突變雜合體(指病變的β珠蛋白基因的核苷酸序列上同時有2種突變點)的雜合子狀態,如[TATAbox-28(A→G)CDl7(A→T)/N],主要表現為輕度小細胞,低色素貧血,尤其嬰兒期明顯,可有輕度黃疸和脾大.

小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血
小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血症状
患兒出生時無癥狀,多於嬰兒期發病,生後3~6個月內發病者占50%,偶有新生兒期發病者,發病年齡愈早,病情愈重,嚴重的慢性進行性貧血,需依靠輸血維持生命,3~4周輸血1次,隨年齡增長日益明顯,伴骨骼改變,首先發生於掌骨,再至長骨,肋骨,最後為顱骨,形成特殊面容(Downs面容):頭大,額部突起,兩顴略高,鼻梁低陷,眼距增寬,眼瞼水腫,皮膚斑狀色素沉著,食欲不振,生長發育停滯,肝脾日漸腫大,以脾大明顯,可達盆腔,患兒常並發支氣管炎或肺炎,並發含鐵血黃素沉著癥時因過多的鐵沉著於心肌和其他臟器如肝,胰腺等而引起該臟器損害的相應癥狀,其中最嚴重的是心力衰竭和肝纖維化及肝功能衰竭,是導致患兒死亡的重要原因之一,本病如不治療,多於5歲前死亡.
小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血
小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血检查
1.血象
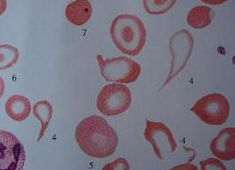
Hb100~120g/L,紅細胞<2.0×1012/L,紅細胞大小不等,呈小細胞低色素性貧血,中央淺染,外周血塗片紅細胞異形明顯,可見梨形,淚滴狀,小球形,三角形,靶形及碎片,嗜堿性點彩紅細胞,多嗜性紅細胞,有核紅細胞增加,網織紅細胞增加(≤0.1),白細胞及血小板數增加,並發脾功能亢進.
2.骨髓象
有核紅細胞增生極度活躍,粒∶紅比值倒置,以中,晚幼紅細胞為主,胞體小,核固縮,胞漿少偏藍,甲基紫染色可見晚幼紅細胞含包涵體(僅鏈沉淀).
3.紅細胞鹽水滲透性試驗
紅細胞滲透脆性減低,0.3%~0.2%或更低才完全溶血.
4.HbF測定
這是診斷重型β珠蛋白生成障礙性貧血的重要依據,HbF含量輕度升高(<5%)或明顯增高(20%~99.6%);HbA2常降低,正常或中度增高,HbA23.5%~8.0%.
5.肽鏈分析
采用高效液相層析分析法可測定α,β,γ,δ肽鏈的含量,Cooley貧血時,β/α比值<0.1(正常值為1.0~1.1),因本病多為點突變,故常用PCR加ASO才能明確突變點,我國各民族的β地貧基因突變情況有一定差異,南方漢族的突變基因以CD41-42(-TCTTT),CDL7(A→T),IVS-Ⅱ-654(C→T)和TATA-box28(A→G)為主,占85%~90%,雙重雜合子的突變組合可達近100種.
6.其他
血漿及紅細胞內維生素E含量顯著下降,與病情呈正相關;超氧陰離子自由基增加.
常規做X線,B超,心電圖等檢查.
骨骼X線檢查,骨髓腔增寬,皮質變薄和骨質疏松,顱骨的內外板變薄,板障加寬和短發樣骨刺形成.
小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血预防
積極開展優生優育工作,以減少/控制“地中海貧血"基因的遺傳.
1.婚前地中海貧血篩查,避免輕型地中海貧血患者聯婚,可明顯降低重型/中間型地貧患者出生的機會.
2.推廣產前診斷技術,對父母雙方或一方地貧基因攜帶者,孕4個月時,采集胎兒絨毛,羊水細胞或臍血,獲得基因組DNA以聚合酶鏈反應(PCR)技術對高危胎兒進行產前診斷,重型/中間型患兒應終止妊娠.
小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血治疗
1.治療原則
輕型地中海貧血不需治療;中間型α地中海貧血應避免感染和用過氧化性藥物,中度貧血伴脾腫大者可做切脾手術,中間型β地中海貧血一般不輸血,但遇感染,應激,手術等情況下,可適當予濃縮紅細胞輸註;重型β地中海貧血,高量輸血聯合除鐵治療是基本的治療措施;造血幹細胞移植(包括骨髓,外周血,臍血)是根治本病的惟一臨床方法,有條件者應爭取盡早行根治手術.
2.輸濃縮紅細胞
(1)低量輸血:單純的輸血或輸紅細胞最終導致血色病,中等量輸血療法,使血紅蛋白維持在60~70g/L,實踐證明,這種輸血方法雖然使重型患者有望擺脫近期死亡的威脅,但患者的生存質量隨年齡增長越來越差,相當一部分患者於第2個十年內因臟器功能衰竭而死亡.
(2)高量輸血:①高量輸濃縮紅細胞的優點:糾正機體缺氧;減少腸道吸收鐵;抑制脾腫大;糾正患兒生長發育緩慢狀態,②方法:先反復輸濃縮紅細胞,使患兒血紅蛋白含量達120~140g/L,然後每隔3~4周Hb≤80~90g/L時輸註濃縮紅細胞10~15ml/kg,使Hb含量維持在100g/L以上.
3.鐵螯合劑
因長期高量輸血,骨髓紅細胞造血旺盛,“無效紅細胞生成"以及胃腸道鐵吸收的增加,常導致體內鐵超負荷易合並血色病,損害心肝,腎及內分泌器官功能,當患者體內的鐵累積到20g以上時,則可出現明顯的中毒表現,故應予鐵螯合劑治療,1歲內使用鐵螯合劑,其副作用如骨骼畸形,生長抑制的發生率明顯升高,一般主張2~3歲後或患兒接受10~20次輸血後並有鐵負荷過重的證據,血清鐵(SF)>1000μg/L,血清轉鐵蛋白完全飽和才開始除鐵治療,當前臨床廣泛使用的是去鐵胺(Deferoxamine,DFO),劑量:20~50mg/(kg·d),加註射用水或生理鹽水用便攜式輸液泵每天(或每晚)腹壁皮下註射8~12h,每周連用5~6天,用藥前後應作SF,尿鐵的監測,若SF>3000μg/L或者有鐵負荷繼發心臟病時,可予DFO50~70mg/(kg·d)持續24h靜脈滴註,使用鐵整合劑時加用維生素C口服可增加尿中鐵的排泄量1倍,但維生素C可將鐵從儲備部位動員出來並通過氧化代謝間接影響心肌細胞,故在重度鐵負荷時不宜使用大劑量維生素C,一般每天口服100~200mg,在停用DFO期間也不應堅持服維生素C,長期使用DFO一般無明顯的毒副作用,註射局部反應,皮疹,疼痛,無需停藥,但鐵負荷輕者使用大劑量DFO可出現白內障,聽力喪失,長骨生長障礙等,應引起臨床重視,Johon等對47例地貧患者接受DFO治療的毒副作用研究發現,DFO大劑量與SF<2000μg/L是引起DFO毒性的兩大危險因素,提出治療指數(TI),即平均每天DFO劑量(mg/kg)除以血清鐵蛋白濃度(μg/L),可指導臨床給藥,當TI<0.025時,一般無毒性,近十年來,國外一系列新型口服鐵螯合劑如defefipone(L1),多價陰離子胺(thepolyanionicanine,HBED),多價氮替代物(thesubstitutedpolyazacompounoxTRcoll),PIH等相繼問世,在動物實驗中已證實長期服用能有效地降低機體鐵負荷,但隻有L1試用於人體,通過高量輸血與除鐵治療可維持患者正常生長發育及達到正常人的生活質量及壽命,但必須終身承受沉重的經濟負擔,可能的輸血相關合並癥及心理負擔.
4.造血幹細胞移植(HSCT)HSCT
是當前臨床上根治本病的惟一方法,HSCT包括骨髓移植(BMT),臍血移植(UCBT),外周血造血幹細胞移植(PBSCT)和宮內造血幹細胞移植(IUSCT),迄今,全世界已成功開展HSCT1200例,其中BMT達1000餘例,PBSCT10例,UCBT約30例,IUSCT2例,研究發現,重型地貧的HSCT有其自身的特點.
(1)受體的選擇:移植前病者3個危險因素評分標準將其分為3類:Ⅰ類:零分,Ⅱ類:1~2分,Ⅲ類:3分,危險因素評分:①去鐵胺應用史:A.“0"分為規則使用,即經1次規則輸血後18個月開始,每周至少5天,皮下輸註持續8~10h,B.“1"分為不規則使用,未達上述任一標準,②肝大:A.“0"分為肝活檢無纖維化,B.“1"分為纖維化,BMT效果順序為Ⅰ>Ⅱ>Ⅲ類;無病存活率分別為85%,80%,53%,纖維化及鐵負荷是重要危險因素.
(2)供體選擇:①血緣相關HLA全相合的供體:同胞HLA相合供體BMT可治愈80%的重型β地貧,但僅25%~30%患者可找到HLA相合的傢系成員供體,作者等自1998年1月國內首例親屬UCBT治療地貧取得成功以來,至今共完成HSCT12例,植入率83.3%,5年無病存活率(EFS)58.3%,②血緣相關HLA不全相合(包括同胞,雙親)或單倍體相合供體:Polchi等報道18例患者BMT後生存率及無病生存率分別為58%和26%,排斥率41%,總病死率44%,Gaziev等報道29例結果:植入率44.8%;排斥率55%,相關病死率34%,因此,找不到HLA相合供體,且無法接受輸血及去鐵治療者,才考慮進行此類移植,③非血緣相關(UD)供體:Dihi報道3例地貧UD-BMT:1例植活,1例回復地貧狀態,1例死於移植物抗宿主病(GVHD);Miano報告8例,4例成功,3例排斥,1例死亡,發生排斥是此類移植面臨的主要問題,2000年6月,作者等以非血緣相關UCBT治療1例重型地貧患兒,取得瞭成功,現患兒已脫地貧狀態存活180天,IUSCT近年已有成功報道,1996年Touraine等用胎肝造血幹細胞宮內移植治療2例宮內確診為地貧胎兒,1例死亡,1例生下無病生存已達4年之久,但Monni等用父親骨髓CD34細胞經胎兒腹腔註射治療1例胎齡為10周的重型B地貧胎兒未獲成功,目前IUSCT成功所需單個有核細胞數,移植的最佳胎齡,植入後的狀態等尚需進一步深入研究.
5.脾切除,大部分脾栓塞術
(1)重型β地中海貧血:①脾切除指征:重型β地貧伴脾功能亢進者,行脾切除能減少對輸血的需要量,並減輕體內鐵負荷,但切脾僅是姑息療法,而且,切脾面臨著發生嚴重感染等致命並發癥的危險,故脾切除應有嚴格的指征:A.輸血量B.脾功能亢進:紅細胞破壞增加,持續的白細胞或血小板減少,C.巨脾引起壓迫癥狀,D.一般年齡應在5歲以上,Maniga等提出輸血商(TQ)患者每年每公斤體重的輸血量除以同齡維持同一Hb水平,脾臟正常患者每年每公斤體重的輸血商>1,可作為臨床判斷早期脾功能亢進的指標,②脾大部分栓塞治療(PSE):由於脾臟為重要的免疫器官,為避免脾切除後繼發性免疫功能低下和兇險的感染,有學者提出脾大部分栓塞治療(PSE)代替脾切除,Politis等對比瞭PSE與脾切除術後5年的追蹤隨訪資料,結果顯示兩組病人輸血量較前都有減少,但PSE組血中IgM濃度明顯高於脾切除組,且感染的發生率也明顯低於後者,但有資料顯示PSE對中間型β地貧效果較滿意,對重型β地貧的療效不滿意,可能與重型β地貧粗大顆粒的紅細胞包涵體主要在骨髓破壞有關,以上2種手術後均可導致肝臟代償性溶血,引起明顯肝大,纖維化加重.
(2)中間型僅地中海貧血(HbH)病:中度/重度貧血(Hb<80g/L)無黃疸的HbH患病者,行切脾術療效極佳,可使Hb上升至90~110g/L,若術前Hb>80g/L或慢性溶血性黃疸者,切脾常無效.
6.基因治療
從分子水平上糾正致病基因的表達,即基因治療,其途徑有2:
(1)將正常的β珠蛋白的基因導入患者的造血幹細胞,以糾正β地貧的遺傳缺陷,必須解決以下3個難題:①轉移的外源珠蛋白基因能在細胞和整體達到高度表達,②必須分離,純化獲得用於基因傳導的人類造血幹細胞,③α基因與β基因之間表達協同一致性,此外,轉導的外源性基因必須隨珠蛋白基因系統在個體發育過程中適時表達仍處於實驗研究階段.
(2)采用某些藥物調節珠蛋白基因的表達:采用某些藥物調節珠蛋白基因的表達以平衡α,β珠蛋白的肽鏈水平,目前臨床應用以調節珠蛋白基因表達的藥物有白消安(馬利蘭),羥基脲,丁酸鹽,阿糖胞苷(Ara-C),紅細胞生成素(Epo)和異煙肼(雷米封)等,其中羥基脲(Hu)應用及實驗研究較多,羥基脲(Hu)是一種低毒和可有效增加γ珠蛋白鏈和β珠蛋白鏈合成,從而導致血液學和臨床癥狀明顯改善,羥基脲(Hu)治療的劑量及方法:①5天療法:50mg/(kg·d)×5天為1個療程,②10~30mg/(kg·d),連用3周為1個療程,或25~50mg/(kg·d)×5~7天為1個療程,③有學者采用15~20mg/(kg·d)連續用藥方法,主要對某些β地貧基因缺陷類型有效:A.-28/654-2或-28/41-42雙重雜合子,β-28純合子,B.IVS-2-654C→T突變中間型β-地貧,C.HbE/β-28雙重雜合子,5~7天顯效,Hb上升水平20~45g/L,中間型效果明顯,重癥者一般用藥初期效果明顯,隨治療時間延長,效果漸差,人們正對這種藥物調控基因的機制深入研究,從而加深對珠蛋白基因表達的遺傳調控的認識.
小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血饮食
1.高蛋白低脂肪:對一般貧血病人來說,首先應考慮給予高蛋白飲食.這可以通過食用動物的瘦肉以及肝,腎等內臟,獲得優質蛋白的補充.其次,應盡量控制脂肪的攝入量,因為脂肪可抑制人體的造血功能,高脂肪還可導致腹瀉、消化不良、肥胖病等疾患.
2.豐富的維生素:飲食結構中維生素的含量豐富,對各類疾病的患者都是適宜的.就貧血病人而言,維生素B1,維生素B12,維生素C和葉酸等是至關重要的.維生素B1的補充,可以通過糧食特別是粗雜糧食物獲得;維生素B12和葉酸主要來源於動物內臟等食物;維生素C的主要來源則是各種新鮮的蔬菜和水果.
小兒β地中海貧血 海洋性貧血 珠蛋白生成障礙貧血并发症
可並發水腫、腹水、貧血、骨骼改變,生長發育停滯,常並發支氣管或肺炎,並發含鐵血黃素沉著癥,造成臟器損害,並發心力衰竭、肝纖維化、肝功衰竭等.具體如下:
1、黃疸:常見癥狀與體征,其發生是由於膽紅素代謝障礙而引起血清內膽紅素濃度升高所致.臨床上表現為鞏膜、黏膜、皮膚及其他組織被染成黃色.
2、肝脾腫大:肝臟和脾臟均增大.肝脾一般在肋下不能觸及,當內臟下垂或橫膈下降或深吸氣時,肝脾才能被觸及,但不超過肋下1cm,且質地較軟.在長期貧血和溶血的刺激下,不少重型和中型貧血病者都會出現脾臟發大的問題.過大脾臟會使貧血加劇和令病者需要接受更大量的輸血而導致更嚴重的鐵質積聚.及時把發大的脾臟切除往往能令情況改善.
3、溶血危象:可有嚴重的腰背及四肢酸痛,伴頭痛、嘔吐、寒戰,隨後面色蒼白和黃疸,是由於溶血產物對機體的毒性作用所致,更嚴重者有周圍循環衰竭.由於溶血產物引起腎小管細胞壞死和管腔堵塞,最終導致急性腎功能衰竭.
4、過量鐵質積聚:長期輸血會造成鐵質沉積而過量鐵質的積聚會對多個器官造成破壞.主要受影響的包括心臟、肝臟、胰臟和各個內分泌器官.病者會出現心臟衰竭、肝硬化、肝功能衰退、糖尿以及因為多種內分泌失調而變得身材矮小和發育不全等等.需註意,除鐵藥有時亦會影響視力、聽覺和骨骼生長.因此,除鐵藥物的註射份量應根據鐵質積聚的多少而定,切勿擅自把份量增加或減少.
5、輸血:其不良反應包括發熱、發冷和出紅疹等.較嚴重的反應如急性溶血、氣管收縮和血壓下降等.另外,經輸血而傳染的主要是過濾性病毒疾病.輸血過程嚴防感染丙型和乙型肝炎甚至愛滋病毒的傳染
6、膽石的形成:長期溶血令地中海貧血病人比一般人更容易患膽石.患有膽石的病人可能經常出現右上腹痛、皮膚、眼白變黃和茶色小便等的病徵.
1/2 1 2 下一页 尾页


















