線粒體病
線粒體病百科
線粒體病是遺傳缺損引起線粒體代謝酶缺陷,致使ATP合成障礙、能量來源不足導致的一組異質性病變.線粒體腦肌病的不同類型發病年齡不同.線粒體是密切與能量代謝相關的細胞器,無論是細胞的成活(氧化磷酸化)和細胞死亡(凋亡)均與線粒體功能有關,特別是呼吸鏈的氧化磷酸化異常與許多人類疾病有關.根據線粒體病變部位不同可分為:1.線粒體肌病線粒體病變侵犯骨骼肌為主.2.線粒體腦肌病病變同時侵犯骨骼肌和中樞神經系統.
線粒體病
線粒體病病因
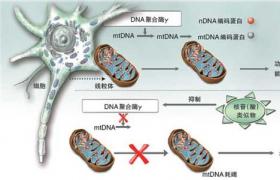
(一)發病原因線粒體是細胞內提供能量的細胞器,人類mtDNA是長16569bp的環狀雙鏈分子,分輕鏈和重鏈,含37個基因,主要編碼呼吸鏈及與能量代謝有關蛋白.mtDNA缺失或點突變使編碼線粒體氧化代謝過程必需的酶或載體發生障礙,糖原和脂肪酸等不能進入線粒體充分利用和產生足夠的ATP,導致能量代謝障礙和產生復雜的臨床癥狀.
(二)發病機制線粒體是密切與能量代謝相關的細胞器,無論是細胞的成活(氧化磷酸化)和細胞死亡(凋亡)均與線粒體功能有關,特別是呼吸鏈的氧化磷酸化異常與許多人類疾病有關.
由於受精卵線粒體均來自卵子,故線粒體病是與孟德爾遺傳不同的母系遺傳方式.與常染色體遺傳病類似,但每一代發病個體多於常染色體遺傳病.母親將mtDNA傳遞給子代,隻有女兒可將mtDNA傳遞給下一代.因每個細胞mtDNA有多重拷貝,線粒體編碼基因表現型與細胞內突變型與野生型mtDNA的相對比例有關,隻有突變型達到某一閾值時患者才會出現癥狀.
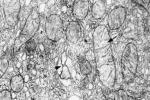
病理變化:肌肉冰凍切片用改良的Gomori三色染色活檢,光鏡下可見異常線粒體聚集的蓬毛樣紅纖維(raggedredfiber,RRF),電鏡顯示大量異常線粒體糖原和脂滴堆積,線粒體嵴排列紊亂.線粒體是密切與能量代謝相關的細胞器,無論是細胞的成活(氧化磷酸化)和細胞死亡(凋亡)均與線粒體功能有關,特別是呼吸鏈的氧化磷酸化異常與許多人類疾病有關.
Luft等(1962)首次報道一例線粒體肌病,生化研究證實為氧化磷酸化脫耦聯引起.Anderson(1981)測定人類線粒體DNA(mtDNA)全長序列,Holt(1988)首次發現線粒體病患者mtDNA缺失,證實mtDNA突變是人類疾病的重要病因,建立瞭有別於傳統孟德爾遺傳的線粒體遺傳新概念.
根據線粒體病變部位不同可分為:
1.線粒體肌病(mitochondrialmyopathy)線粒體病變侵犯骨骼肌為主.
2.線粒體腦肌病(mitochondrialencephalomyopathy)病變同時侵犯骨骼肌和中樞神經系統.
3.線粒體腦病病變侵犯中樞神經系統為主.
線粒體病的癥狀
線粒體病
線粒體病症状
1.線粒體肌病(mitochondrialmyopathy)多在20歲時起病,也有兒童及中年起病,男女均受累.臨床特征是骨骼肌極度不能耐受疲勞,輕度活動即感疲乏,常伴肌肉酸痛及壓痛,肌萎縮少見.易誤診為多發性肌炎、重癥肌無力和進行性肌營養不良等.
2.線粒體腦肌病(mitochondrialencephalomyopathy)包括:
(1)慢性進行性眼外肌癱瘓(CPEO):多在兒童期起病,首發癥狀為眼瞼下垂,緩慢進展為全部眼外肌癱瘓,眼球運動障礙,雙側眼外肌對稱受累,復視不常見;部分病人有咽肌和四肢肌無力.
(2)Keams-Sayre綜合征(KSS):20歲前起病,進展較快,表現CPEO和視網膜色素變性,常伴心臟傳導阻滯、小腦性共濟失調、CSF蛋白增高、神經性耳聾和智能減退等.
(3)線粒體腦肌病伴高乳酸血癥和卒中樣發作(MELAS)綜合征:40歲前起病,兒童期發病較多.表現突發的卒中樣發作,如偏癱、偏盲或皮質盲、反復癲癇發作、偏頭痛和嘔吐等,病情逐漸加重.CT和MRI可見枕葉腦軟化,病灶范圍與主要腦血管分佈不一致,常見腦萎縮、腦室擴大和基底核鈣化;血和腦脊液乳酸增高.
(4)肌陣攣性癲癇伴肌肉蓬毛樣紅纖維(MERRF)綜合征:多在兒童期發病,主要表現肌陣攣性癲癇、小腦性共濟失調和四肢近端無力等,可伴多發性對稱性脂肪瘤.
3.線粒體腦病(mitochondrialencephalopathy)包括Leber遺傳性視神經病(LHON)、亞急性壞死性腦脊髓病(Leigh病)、Alpers病及Menkes病等.
線粒體肌病的診斷依賴於典型的臨床癥狀:四肢近端極度不能耐受疲勞、身體矮小和神經性耳聾等,伴各亞型臨床特征;血清乳酸、丙酮酸增高和肌肉活組織檢查發現RRF,mtDNA缺失或點突變等之結果.線粒體腦肌病患者CT或MRI檢查可見白質腦病、基底核鈣化、腦軟化、腦萎縮和腦室擴大等.
但應註意炎癥肌病和其他肌病可同時伴存線粒體和糖原堆積之可能.嚴防過多過濫診斷線粒體肌病.
線粒體病
線粒體病检查
線粒體病的檢查化驗1.血生化檢查①血乳酸、丙酮酸最小運動量試驗:約80%的病人運動後10min血乳酸和丙酮酸仍不能恢復正常,為陽性;線粒體腦肌病患者CSF乳酸含量也增高;②線粒體呼吸鏈復合酶活性降低.
2.mtDNA分析①CPEO和KSS為mtDNA片斷缺失,可能發生在卵子或胚胎形成期;②80%的MELAS病人為mtDNA、tRNA基因3243點突變;MERRF綜合征是tRNA基因位點8344點突變.
1.肌肉冰凍切片Gomori染色活檢可見肌細胞內線粒體堆積,RRF和糖原脂肪增多.
2.CT或MRI檢查線粒體腦肌病患者可見白質腦病、基底核鈣化、腦軟化、腦萎縮和腦室擴大等.
3.肌電圖可表現為肌源性損害或神經源性損害.
線粒體病的鑒別診斷線粒體肌病需與多發性肌炎、重癥肌無力、周期性癱瘓和眼咽型進行性肌營養不良等鑒別.
線粒體病预防
一、預防遺傳病治療困難,療效不滿意,預防顯得更為重要.預防措施包括避免近親結婚,推行遺傳咨詢、攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生.
二、護理
線粒體病治疗
線粒體病的預防和治療方法遺傳病治療困難,療效不滿意,預防顯得更為重要.預防措施包括避免近親結婚,推行遺傳咨詢、攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生.
線粒體病的西醫治療(一)治療目前無特效治療.可給予三磷腺苷(ATP)、輔酶Q10和大量B族維生素等,丙酮酸羧化酶缺少的患者推薦高蛋白、高碳水化物和低脂肪飲食.部分病例對腎上腺皮質激素反應良好.
(二)預後不同類型預後不同,病程長短不一(參見臨床表現).對癥治療可提高患者生活質量.
線粒體病饮食
一、飲食1、線粒體病吃那些食物對身體好:
飲食療法飲食治療可減少內源性毒性代謝產物的產生.高蛋白、高碳水化合物、低脂飲食能代償受損的糖異生和減少脂肪的分解.
以上資料僅供參考,詳細請咨詢醫生
線粒體病并发症
線粒體病的並發癥隨病情發展,可以出現多樣的癥狀體征(參見臨床表現).
1/2 1 2 下一页 尾页


















