
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病百科
蕈樣真菌病(mycosisfungoides,MF)是一種T細胞起源的惡性腫瘤.又稱蕈樣肉芽腫(granulomafungoides),是一種向上皮性皮膚淋巴瘤.其特征為輔助T細胞增生,Langerhasns細胞和交指狀網狀細胞也參與病變.病程呈慢性漸進性,初期為多種形態的紅斑和浸潤性損害,以後發展成腫瘤,晚期可累及淋巴結合內臟.
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病病因
一、發病原因
大多數病例均為記憶輔助T細胞腫瘤.病因尚不明.以往普遍認為MF從一開始就是惡性腫瘤,但近年來愈來愈多的人認為本病開始時為一免疫性疾病,以後才發展為淋巴瘤.從血管免疫母細胞淋巴結病經常進展為免疫母細胞淋巴瘤這一事實證明瞭此種發展的可能性.此外以下的觀察亦支持MF的免疫學起源:①正常人類淋巴細胞與高陸核絲分裂原或植物血凝素培養引起5%~11%細胞的外觀與光鏡下和電鏡下所見的MF細胞或seary細胞不能區別,說明後者為受刺激的淋巴細胞的產物.②MF中的向表皮性(epidermotropism)和形成Pautrier微膿腫亦代表一種免疫現象,Langerhans細胞提呈抗原給T淋巴細胞,像接觸性皮炎一樣,導致淋巴細胞與Langerhans細胞並位.T淋巴細胞與Langerhans細胞交互作用,從而形成Pautrier微膿腫灶.此種過程比接觸性皮炎慢這一事實提示其在處理抗原方面有缺陷,使一種尚未證實的抗原持續存在,並刺激淋巴細胞在慢性反應過程中發生惡變.③鑒於細胞浸潤的單克隆性(momoclonalily)為惡性的標記,T細胞受體基因克隆重排南方斑點分析(southernblottinganalysis)表明T細胞的單克隆性在早期斑塊期並不明顯,但在硬結性斑塊進展為腫瘤期時,則可查到.此外病毒感染亦被涉及,有報告在315例MF患者中,發現36例(11.4%)有抗人類T細胞親淋巴病毒1型(HTLV-1)的特殊抗體.
二、發病機制
發病機制還不十分清楚,但對本病的發病機制提出以下的假說.本病的皮膚浸潤經過3個階段:
1.不典型皮膚浸潤階段雖然對本病的最初組織病理變化仍有爭論,認為一開始為相互作用的T淋巴細胞和單核細胞的不典型浸潤,以後可消退,局部持久存在或受不同影響發展成下個階段.
2.皮膚擴展階段上述不典型浸潤細胞可以影響再循環T細胞,當後者進入其他部位皮膚時,引起同樣的不典型相互作用或激發最初浸潤的因素直接影響其他部位的皮膚,發生不典型多形性浸潤.隨著病變的發展,形成斑塊或腫瘤時,浸潤變得更單一,主要為T細胞性瘤細胞,甚至在此期已有極輕微的系統性免疫異常.
3.系統性病變階段當皮損發展或累及血液、其他內臟時,單一形T細胞性瘤細胞浸潤突出.患者細胞免疫明顯異常.異常免疫反應可發生在各個方面,可因遊走和再循環的異常方式、尚未明確的皮膚刺激的傳遞(持續性抗原、環境中變應原)、異常T淋巴細胞的反應或輔助或抑制成分的異常調節而在組織中發生淋巴細胞和巨噬單核細胞的聚集.MF的發生與環境因素(如接觸石油工業品、工業廢物、廢氣、放射性污染物、殺蟲劑、農藥以及其他變應原,如光線或病毒)、機體易感性、異常淋巴細胞單核細胞或淋巴細胞Langerhans細胞相互作用,或異常抗原持續性刺激均有關系.根據上述假設和推理,他認為本病在早期為反應性,而後發展成惡性新生物性.
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病症状
一、癥狀
MF原發於皮膚,但最終淋巴結和內臟常受累.
1、臨床表現和組織病理分期典型病例在臨床上和組織病理上可分3期,即紅斑期、斑塊期和腫瘤期.3期可互相重疊,因而3期損害可同時出現.
(1)紅斑期:皮疹呈斑片狀,通常扁平面不萎縮,但有些患者則表現萎縮.扁平非萎縮型斑片常有鱗屑附著,類似銀屑病或濕疹,後者呈圓形、卵圓形,亦可呈環狀、多環狀或弓形.萎縮型斑片表面光亮,易皺縮,正常溝嵴消失,毛細血管擴張,色素減退或色素增深.臨床上.呈血管萎縮性皮膚異色癥,大斑塊型副銀屑病或斑駁性副銀屑病.扁平非萎縮性斑片通常數月數年後繼發浸潤,也可出現內臟損害,而扁平萎縮性斑片則隻有12%的患者演變為侵襲性MF,其餘患者保持現狀無改變.
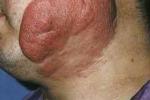
(2)斑塊期:由紅斑期進展而來,或在正常皮膚上發生,呈不規則性,界限清楚略高起的斑塊,顏色黃紅色、暗紅色至紫紅色不等.可自行消退,亦可融合為大的斑塊,邊緣呈環狀、弓形或匍行性,顏面受累時褶皺加深形成“獅面".斑塊可互相融合成廣泛性,但有散在的正常皮膚存在.損害進一步發展可發生疼痛性表淺潰瘍.此期常發生淋巴結腫大,無觸痛,性質堅實,可自由推動.
(3)腫瘤期:可發生於原有斑塊上或正常皮膚上.皮損為大小不等,形狀不一的褐紅色高起結節,傾向早期破潰,形成深在性卵圓形潰瘍,基底被覆壞死性淡灰白色物質,潰瘍邊緣卷曲,好發於軀幹部,但亦可發生於任何部位,甚至口腔和上呼吸道,一旦腫瘤發生,患者通常在數年內死亡.此外MF可見紅皮病型亞類,呈全身性剝脫和皮膚潮紅,毛發稀少,甲營養不良,掌蹠角化,有時全身性色素增深.偶亦見毛囊性黏蛋白病,色素減退損害與MF伴發.
MF為一種惡性淋巴瘤,除皮膚外,淋巴結受累最常見,其他依次為脾、肺、肝、骨髓、腎、舌或會厭、心臟和甲狀腺.內臟受累的患者存活期約1年.
二、MF分期標準
(1)T指皮膚受累:
T1皮膚受累低於10%.
T2皮膚受累高於10%.
T3腫瘤.
T4紅皮病.
(2)N指淋巴結受累:
N0淋巴結臨床上及病理學上均末受累.
N1淋巴結腫大,但病理學上為非MF.
N2淋巴結不腫大,但病理學上為MF.
N3淋巴結腫大,病理學上為MF.
M0內臟未受累.
M1內臟受累.
(3)MF分期如下:
ⅠA為T1,N0,M0.
ⅠB為T2,N0,M0.
ⅡA為T1~2,N1,M0.
ⅡB為T3,N0~1,M0.
ⅢA為T4,N0,M0.
ⅢB為T4,N1,M0.
ⅣA為T1~4,N2~3,M0.
ⅣB為T1~4,N0~3,M1.
主要根據臨床上的特點與組織學的指征,早期診斷一般均需作活檢確定.因此當臨床上懷疑為本病時,應及時做活檢,且往往需連續切片方能找到特異性病變.Farber曾提出用“削片法"連續切片觀察表皮病變.我們建議早期淺表病變可用銳利的刀片僅削取病變處的表皮,連續切片尋找Pautrier微膿腫,同時作細胞塗片檢查.此法比較簡單,對患者損傷較少,不需要縫合,有利於反復取材.但即使如此,有時仍不易確診,故早期診斷方法仍有待於進一步研究.因此對本病診斷應慎重,應臨床與病理及免疫組化結果密切結合,必要時密切隨訪,多次取材,切不可主觀片面,草率從事.目前,下列診斷指征可供參考:
三、臨床方面
(1)皮疹多形,而且同時存在,以致臨床上往往既像某一病,又像另一病;或者既不完全符合某一病,也不完全符合另一病.換言之,這些皮疹表現很難用一種皮膚病來解釋或概括.例如在一患者身上有些損害類似皮膚異色癥,而另一些損害又像魚鱗病.如為紅斑損害,則常帶暗紅色或棕紅色.既增生肥厚,又伴有萎縮.而且常有色素異常、色素沉著與色素減退並存.皮疹邊緣不規則,形態也不一致,分佈又無規律.這些都是本病的特點.
(2)雖然瘙癢在本病開始或病程中並非必有的癥狀,但不少病例均有瘙癢.特別是皮疹泛發而有頑固性劇癢,用一般藥物難以控制者,應懷疑有無本病的可能性
(3)本病雖然部分皮疹可以自然消退,但總的來看,皮疹不斷增多,浸潤逐漸加重,因此呈慢性進行性的過程,病程往往較長,也是其特點之一.
四、病理方面
(1)親表皮現象:真皮內浸潤細胞往往侵入表皮甚至毛囊上皮,即所謂親表皮現象,侵入表皮的單一核細胞,周圍有暈,並有聚集形成Pautrier微膿腫的趨勢.這種現象為本病診斷的重要依據之一.這種親表皮現象與一般濕疹、皮炎所見的細胞外移不同,後者往往伴有海綿水腫,細胞多為中性粒細胞、淋巴細胞混雜在一起,而MF則無明顯水腫,全部為單一核細胞.
(2)MF細胞:斑塊期開始真皮內即可出現相當比例的MF細胞,核深染、形態不規則,甚至大小不一,細胞周圍有透明暈,對診斷有價值.
(3)浸潤形態:呈T細胞模式.早期浸潤多限於真皮上部,多呈帶狀,伴有基底細胞液化,類似皮膚異色病的表現者,往往要考慮本病.
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病检查
一、檢查
1、血象早期血紅蛋白正常,晚期可有輕度貧血.偶或為溶血性貧血.有些病例白細胞增加.嗜酸粒細胞和單核細胞增加,淋巴細胞減少,這在泛發性斑塊和腫瘤期患者中尤為常見.提示預後較差.我們的病例大多為Ⅰ~Ⅱ期,27.5%有嗜酸粒細胞增加,47.5%有單核細胞增加,76%有淋巴細胞減少.文獻報道約20%病例(我們70.8%的病例)血中可找到異常淋巴細胞,占有核細胞數的6%~35%,大多在20%以下.
2、骨髓象一般正常,偶見漿細胞增加.我們檢查9例,6例嗜酸粒細胞和漿細胞增加,2例巨噬細胞增生,6例見異常淋巴細胞,占有核細胞數2%~3%.
3、外周血電解質正常,我們測定20例血鈣均正常.尿酸可增加.晚期血清白蛋白減少,α1球蛋白和α2球蛋白增加.循環輔助T細胞減少,對PHA刺激的反應性降低,無效細胞增加,單核細胞趨向性降低,單核細胞抑制因子減少,血清IgG和IgE增加.
4、紅細胞沉降率測定70例,其中60例(80.5%)有不同程度的增快(15~30mm/h).
5、免疫測定①細胞免疫反應(包括結核菌素試驗、DNCB試驗、鏈素反應以及淋巴細胞轉換率測定)呈陰性或低於正常.40例DNCB試驗,其中22例(53.84%)為陰性;50例OT試驗,其中32例(64.28%)為陰性;20例LTT,其中15例(78.58%)低於正常.②熒光檢查示血管壁中有IgG、IgA、IgM和IgD沉積.
6、其他如肝臟累及時,血清堿性磷酸酶值增加,其他肝功能測定也異常.肺臟受累時,X線攝片示腫瘤樣陰影,但無特征性.
1、組織病理
(1)紅斑期:早期診斷困難,常僅在真皮上部見非特異性炎癥浸潤.但即使在早期,也時常可見親表皮現象(epidemotropism),即表皮內出現散在單個的單一核細胞,與周圍角質形成細胞之間有一透明間隔或將其分開.偶亦見幾個單核細胞聚集一起,周圍有一暈樣間隔,提示為小的Pautrier微膿腫.此種親表皮現象,常提示為早期MF,與通常各種皮炎中常見的細胞外滲(exocytosis)不同之處在於蕈樣肉芽腫通常無或很少有海綿水腫.
(2)斑塊期:在大多數病例中,本期組織學有診斷價值.真皮上部出現帶狀多形性細胞浸潤,包括淋巴細胞、組織細胞、嗜酸性粒細胞、漿細胞和相當比例的MF細胞(核深染、外形不規則的T淋巴細胞).在真皮下部也能見到斑片狀浸潤.表皮內出現親表皮現象及pautrier微膿腫為本病有診斷價值的特征.與紅斑期的區別在於斑塊期表皮內的單核細胞,有些是MF細胞,而且不僅在表皮,在附屬器上皮,特別是毛囊也可見散在單個核細胞侵入.
(3)腫瘤期:可見兩種組織學表現.有些患者為類似斑塊的多形性浸潤,但大多數病例中浸潤伸展到皮下脂肪層,表皮可呈典型的親表皮性或不受侵,甚至在真皮上層出現無浸潤帶.另一些患者則浸潤呈單形性,幾乎完全由腫瘤細胞構成,親表皮性已不再是特點.在同一患者身上可見到由親表皮性過渡到非親表皮性兩種組織學表現.
2、提示MF診斷的主要組織學特點基底細胞層有單個或小群淋巴細胞;淋巴細胞親表皮性與表皮海綿形成輕微之間不成比例;MF表皮內淋巴細胞比正常炎癥性皮膚病時所見者更多;表皮內淋巴細胞較真皮內者大;角質層和顆粒層可見淋巴細胞;真皮乳頭層纖維化,膠原束平行排列;淋巴細胞明顯親毛囊性(folliculotropism)特別是伴發毛囊內黏蛋白沉積(毛囊性黏蛋白病follicularmucinosis).
3、提示炎癥性皮膚病診斷的主要組織學特點真皮上部和真皮乳頭水腫;表皮海綿形成明顯;表皮內炎癥細胞旱細頸瓶狀聚集,其頂端開口於角質層.
4、免疫組織化學對診斷MF價值有限.MF細胞以CD4,但喪失CD7抗原為特征,亦即MF為CD4CD7-.此種表現型對非惡性T細胞很少見,因而對評價周周血淋巴細胞是有價值的.免疫組化較難應用於隻有少數腫瘤細胞的早期損害,因為在大多數良性病的炎癥性浸潤中也是由輔助T細胞構成,而早期MF有正常輔助T細胞,因此用免疫組化學檢查並不能對二者進行鑒別.另外,DNA分子雜交或南方斑點試驗常在不能確定的病例中進行檢查,以發現T細胞受體(TCR)的克隆重排,但對這些資料必須小心解釋;克隆並不能證實惡性腫瘤診斷.良性病亦可含有克隆TCR重排.早期MF浸潤細胞的數目對檢查到克隆可能不足,因此陰性結果並不能排除MF診斷.類似的技術亦可應用,以檢查MF患者的淋巴結.淋巴結受累亦可通過這些分子學方法查出,而常規組織學檢查可能正常.較慢性期疾病很可能在其淋巴結中存在克隆,而克隆的存在則預示患者存活時間較短.
5、其他特征性免疫表現型為CD-,CD2,CD3,CD5,CD45Ro,CD8-,CD3-.罕見病例亦有報告表達CD3,CD4-,CD8成熟T細胞表現型者.此外腫瘤期喪失T細胞抗原.
二、鑒別
在鑒別診斷方面,因本病可類似很多皮膚病,故不能一一敘述,可結合臨床與病理所見分別加以排除,必要時需要觀察病程,不同時期多次取材,才能加以鑒別.
電鏡觀察MF細胞的形態,發現此種細胞胞質很少,但有相對大的核,核膜有許多皺折,結果核質形成指狀突起,從立體來看,核呈腦回狀.異染色質顆粒致密聚集在核膜上,並聚集成塊,散佈於整個核內.MF細胞與Szary細胞(甚至大斑片型斑片狀副銀屑病中的浸潤細胞)在電鏡表現上有很大類似之處,故無鑒別診斷意義.為瞭診斷的目的,曾試用電鏡測量皮膚活檢中淋巴細胞的核外形,有報告在早期MF與良性炎癥性皮膚病的平均核外形指數(NCI)等有顯著差異,MF細胞NCI常大於7.1~7.5,從而使95%以上的病例可以區別.
李傑等曾測量13例MF細胞,單項指標為:淋巴樣細胞平均核外廓指數(mNCI)5.66;平均核形態指數(mNSI)0.44;NCI6.0的細胞數30%,最大NCI(NCImax)9.0,而NSI為0的淋巴樣細胞的百分率19%.經電子計算機判斷分析,最大NCI值結合NSI為0的細胞百分率是最佳判斷指標,對早期診斷MF頗有幫助.
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病预防
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病治疗
1、治療
以增強患者免疫力為主.許多療法可使疾病獲得時間不等的緩解.局部皮質類固醇激素治療,局部氮芥治療或卡莫司汀(卡氮芥)和PUVA可用於分期ⅠA,ⅠB和ⅡA.全身皮膚電子束治療可用於分期ⅡA,ⅡB.單劑化療或光免疫化學療法可用於早期治療Ⅲ期患者.系統性化療,維A類,光化學免疫療法和α幹擾素可用於Ⅵ期治療.
1.局部皮質類固醇激素療法早期(斑片期T1和T2)可用高強皮質類固醇乳劑外用.T1患者完全緩解為63%,總有效率為94%.T2患者完全緩解率僅25%,總有效率為82%.治療停止後易復發.
2.局部氮芥療法100mg氮芥溶於60ml自來水中外用全身(陰部除外),每天1次,應用數月.80%ⅠA患者,68%ⅠB患者,61%ⅡA患者,49%ⅡB患者,60%Ⅲ期患者顯效.約10%患者可獲得長期緩解8年以上.主要副作用為皮膚過敏;應用軟膏基質可減輕反應,但療效較水劑差.治療停止後,至少1/2患者復發,再治療仍有效.
3.局部卡莫司汀(卡氮芥)治療配制每毫升含2mg卡莫司汀的乙醇液,每天用此種原液5ml加水至60ml.每天1次外用全身(皺襞部、陰部及手足部如無皮損,可不用).皮損局限時,可僅外用於患部.平均療程8~12周.如經3~6個月治療無效,則可將藥物濃度加倍,重復治療12周.小片及頑固性皮損,可直接用原液外用治療.患者對卡莫司汀(卡氮芥)的耐受性較氮芥好,接觸過敏少見.但應用低濃度卡莫司汀(卡氮芥)患者中,少於10%的患者可發生骨髓抑制.長期應用卡莫司汀(卡氮芥)治療者可發生持久和嚴重的毛細血管擴張.
4.紫外線療法約75%斑片期患者,用UVB治療可完全緩解.PUVA應用更廣,由於其較深的穿透性,更適合真皮內損害.限局性斑片或斑塊損害的患者中,88%的患者損害完全緩解,損害廣泛者,52%患者可完全緩解.腫瘤期患者無效.紅皮病患者對PUVA耐受性差.PUVA治療緩解期短,平均約1年,因此需經常維持治療.亦可用維A類和幹擾素α與PUVA聯合應用.
5.體外光化學療法治療前患者服用補骨脂素,然後將血細胞抽出用PUVA在體外處理.約20%的MF皮損完全緩解,20%患者部分緩解.紅皮病患者總有效率為80%.本療法費用昂貴,應先考慮其他更經濟的治療方法.
6.放射療法全身電子束治療,劑量超過3000Gy對MF的治療很有效.分期T1患者98%可完全緩解;T27l%;T336%;T464%.T1患者50%可長期緩解;T2患者20%可長期緩解.紅皮病型耐受性差.常見副作用為紅斑,水腫,損害加重,禿發和甲脫落.
7.生物反應改變劑α幹擾素對MF治療有效率約60%,19%可完全緩解.可有發熱、寒戰、白細胞減少和抑鬱等毒副作用.幹擾素療法可與維A酸療法聯合應用.
8.維A酸類維A酸(異維甲酸)和阿維A酯(依曲替酯)治療MF44%有效.開始劑量1mg/(kg·d),根據患者耐受劑量可增至3mg/(kg·d).維A酸類對IB(T2)患者,Ⅲ期患者有效.對Ⅳ期患者可減輕癥狀.Targretin為一種合成性維A酸,能與RXR受體優先結合,引起許多腫瘤細胞凋亡,對MF亦有效.
9.系統性化療系統性化療用於晚期MF治療,常用單一制劑.氨甲蝶呤5~125mg/周,對T3患者治療有效.41%的患者可達完全緩解,17%患者部分緩解,總有效率為58%.平均存活率為8.4年,69%患者可存活5年.環磷酰胺、多柔比星(阿黴素)、長春新堿和潑尼松龍(CHOP)聯合化療可用於晚期患者.
10.熔毒療法DAB389IL-2為融合白喉毒素於白介素-2的重組體.此制劑可選擇性地與表達白介素-2受體的細胞接合,導致該細胞死亡.該制劑治療表達白介素-2受體的MF病例,有效率為37%,其中完全緩解者為14%.副作用為發熱、寒戰、低血壓、惡心和嘔吐.大劑量應用時可引起血管滲漏綜合征(vascular-leaksyndrome).
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病饮食
暫無具體資料
蕈樣真菌病 蕈樣黴菌病蕈樣肉芽腫蕈樣肉芽腫病并发症
暫無具體資料
2/2 首页 上一页 1 2


















