肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥百科
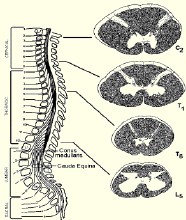
肌張力障礙綜合征(dystonicsyndrome)簡稱肌張力障礙(dystonia),是主動肌與拮抗肌收縮不協調或過度收縮,引起以肌張力異常動作和姿勢為特征的運動障礙綜合征,具有不自主性和持續性特點.盡管名為肌張力障礙綜合征,但肌張力的變化不引人註意,而引人註意的是異常的體位姿勢和不自主的變換動作.肌張力障礙可影響受累肢體的正常運動幅度、范圍、速度和肌肉的硬度等.
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥病因
(一)發病原因
特發性肌張力障礙(idiopathicdystonia)病因不明,可能與遺傳有關,繼發性肌張力障礙常是基底核、丘腦及腦幹網狀結構等病變的癥候.
(二)發病機制
特發性肌張力障礙(idiopathicdystonia)可為常染色體顯性(30%~40%外顯率)、常染色體隱性或X連鎖隱性遺傳,顯性遺傳缺損基因DYT1已定位於9號常染色體長臂9q32~34,編碼ATP結合蛋白扭轉蛋白A(torsinA),可有散發病例.環境因素如創傷或過勞等可誘發,如口-下頜肌張力障礙發病前,可有面部或牙損傷史,一側肢體過勞,常誘發書寫痙攣、打字員痙攣和運動員肢體痙攣等.
繼發性肌張力障礙是紋狀體、丘腦、藍斑、腦幹網狀結構等病變所致,如肝豆狀核變性、膽紅素腦病、神經節苷脂沉積癥、蒼白球黑質紅核色素變性、進行性核上性麻痹、特發性基底核鈣化、甲狀旁腺功能低下、中毒、腦卒中、腦外傷、腦炎等;另外,藥物(左旋多巴、吩噻嗪類、丁酰苯類、甲氧氯普胺)也可誘發.
肌張力障礙還可由心理因素引起,特點是易受到暗示影響.
病理及神經生化改變:特發性扭轉痙攣可見非特異性病理改變,包括殼核、丘腦及尾狀核小神經元變性,基底核脂質及脂色素增多.繼發性扭轉痙攣病理學特征隨原發病不同而異,痙攣性斜頸、Meige綜合征、書寫痙攣和職業性痙攣等局限性肌張力障礙無特異性病理改變.
拮抗肌過度協同性收縮是本病主要的生理學特點,肢體每次收縮常由近端向遠程擴展.肌電圖檢查發現,肌肉收縮間歇期無不自主運動電位.根據肌電圖特點分為3型:①持續30s的肌肉收縮,間隔短時間靜息;②重復、節律性收縮和靜息狀態,收縮期和靜息期均為1~2秒鐘;③快速、短暫肌肉收縮,持續100ms,表現類似肌陣攣.
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥症状
肌張力障礙的臨床分類目前尚不統一.肌張力障礙的各種分類與臨床特點相關,主要根據肌張力障礙的受累肢體和部位,可能造成肌張力障礙的原因,發病時的年齡等.
1.國內王維治主編的《神經病學》(2006年版)介紹如下分類
(1)按病變累及部位分類:
①局限性肌張力障礙:影響軀體某部,如痙攣性斜頸、痙攣性發音困難(聲帶)、瞼痙攣、口-下頜肌張力障礙、書寫痙攣(一側上肢)、職業性痙攣、足肌張力障礙等.
②節段性肌張力障礙:表現節段性分佈特點,如顱-頸部肌張力障礙,上肢伴或不伴中軸、頭頸部肌張力障礙,下肢伴或不伴軀幹肌張力障礙,軀幹-頸部(不影響頭面部)肌張力障礙等.
③多部位肌張力障礙:累及身體2個以上不靠近部位.
④廣泛性肌張力障礙:影響廣泛的軀體范圍.
⑤偏側性肌張力障礙:僅累及單側身體.
(2)按病因分類:
①遺傳性進行性肌張力障礙:多在小兒發病,表現步行障礙及運動減少,軀幹姿勢異常,腰部明顯向前彎曲,日內癥狀波動明顯,晨輕,午後至傍晚加重,呈進行性發展,L-dopa療效顯著,本病與Parkinson病可能有類似之處.
②癥狀性肌張力障礙:如腦性癱瘓、累及基底核的腦卒中、Wilson病、Hallervorden-Spatz病、腦炎、腦腫瘤、精神病藥物副反應等伴肌張力障礙,Parkinson病有時可伴肌張力障礙.
2.上海史玉泉等主編的《實用神經病學》(2004年版)則主張以下分類
(1)按肌張力障礙范圍的分類:
①局限性肌張力障礙(focaldystonia):僅累及眼瞼部肌群者,稱眼瞼痙攣(blepharospasm);累及口周及下頜肌群者,則稱為口下頜肌張力障礙(oromandibulardystonia);疾病累及喉部肌群稱為痙攣性構音障礙(spasmodicdysphonia);疾病累及頸部肌群稱為痙攣性斜頸(spasmodictorticollis);疾病累及前臂和手部,稱為書寫性痙攣癥(writercramp).
②節段性肌張力障礙(segmentaldystonia):累及顱和頸部兩處肌群者,稱之為顱部節段性肌張力障礙(cranialsegmentaldystonia);累及頸和軀幹者稱為縱軸節段性肌張力障礙(axialsegmentaldystonia);累及一側手臂和縱軸軀幹或雙上臂者,稱為臂部節段性肌張力障礙(brachialsegmentaldystonia);累及一側股肌和軀幹或兩側股肌(可有或無軀幹受累)者稱為股節段性肌張力障礙(cruralsegmentaldystonia).
③偏身肌張力障礙(hemidystonia):系指同側上下肢的肌群受累,多為繼發性原因所造成的.
④全身肌張力障礙(generalizeddystoina):系指疾病累及3個或3個以上肢體伴軀幹、顱、頸或延髓部肌群,如全身性的扭轉痙攣(torsionspasm).
(2)按肌張力障礙起病年齡分類:
①兒童型肌張力障礙:患者年齡小於12歲.
②少年型肌張力障礙:患者年齡在13~20歲.
③成年型肌張力障礙:患者年齡大於20歲.
兒童期起病的肌張力障礙,病情常呈進行性,首先累及下肢,出現步態障礙,以後導致全身性肌張力障礙,預後較差.成年型肌張力障礙常為局限性,如頸、上肢,很少累及全身,腿部不受累,病情不進行性加重.
(3)按肌張力障礙病因分類:
①原發性肌張力障礙:
A.遺傳性(常染色體顯性或隱性遺傳、X性連鎖隱性遺傳):肌陣攣性肌張力障礙(酒精反應性、常染色體顯性遺傳),發作性肌張力障礙(paroxysmaldystonia),發作性睡眠性肌張力障礙(paroxysmalhypnogeneticdystonia),典型“特發性"扭轉型肌張力障礙(單純肌張力障礙、常染色體顯性遺傳、dytl基因),非典型“特發性"扭轉型肌張力障礙(常染色體顯性遺傳),不典型肌張力障礙(常染色體顯性遺傳).
B.散發性肌張力障礙.
②繼發性肌張力障礙:包括多巴反應性肌張力障礙(基因位於染色體14),散發性肌張力障礙伴帕金森綜合征,X-性連鎖隱性遺傳肌張力障礙-帕金森綜合征(菲律賓人中),遺傳性肌陣攣性肌張力障礙,少年型亨廷頓舞蹈病,傢族性肌萎縮性肌張力障礙性截癱,哈勒沃登-施帕茨(Hallervorden-Spatz)病,進行性蒼白球變性,遺傳性共濟失調,肌張力障礙伴神經性聾,約瑟夫(Joseph)病,共濟失調毛細血管擴張癥,Rett綜合征,嬰兒雙側紋狀體壞死,傢族性基底核鈣化,神經棘紅細胞增多癥等.也有酚噻嗪類藥、顱腦外傷、基底核腫瘤、代謝性疾病造成.
3.肌張力障礙的主要臨床特點多於10~20歲發病,初期進展較快,爾後逐漸緩慢,有時不進展或稍改善,病程不定.日本散發病例較多,其他國傢傢族性發病較多.臨床表現癥狀體征多樣,如扭轉痙攣樣運動、痙攣性斜頸、書寫痙攣、投擲運動和姿勢異常,步行姿勢不自然等.初期立位或動作時常不自主出現姿勢或肢體位置異常,臥位時肌緊張消失.隨病程進展,臥位也出現異常肌緊張,可見骨變形或肌肉異常發育,粗大動作時各肌肉肌力雖無明顯異常,但因肌緊張不能完成精巧動作,但仍可寫字、步行和騎自行車,有時可伴手指震顫或肌陣攣動作等.
特發性痙攣性斜頸是頸肌肌張力障礙的表現.書寫痙攣(writercramp)是執筆書寫時手和前臂出現肌張力障礙姿勢,表現握筆如握匕首、手臂僵硬、手腕屈曲、肘部不自主地向外弓形抬起、手掌面向側面等,但做其他動作正常.也可表現其他職業性痙攣,如彈鋼琴、打字,使用螺絲刀或餐刀等.藥物治療通常無效,可讓病人學會用另一隻手完成這些任務.
對扭轉痙攣、痙攣性斜頸、書寫痙攣、投擲運動等肌張力障礙患者較易診斷.確定本綜合征後,還應查找病因.
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥检查
肌張力障礙綜合征的檢查化驗
血電解質、藥物、微量元素及生化檢查,有助於病因診斷及分類.
1.CT或MRI檢查對鑒別診斷有意義.
2.正電子發射斷層掃描(PET)或單光子發射斷層掃描(SPECT)可顯示腦部某些生化代謝情況,對診斷頗有意義.
3.基因分析對確診某些遺傳性肌張力障礙疾病有重要意義.
肌張力障礙綜合征的鑒別診斷
肌張力障礙綜合征須註意與肌陣攣、癔癥等鑒別.本病的肌緊張是中樞性較劇烈的不隨意肌收縮,註意與Parkinson病肌強直不同.
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥预防
有遺傳背景的疾病,預防顯得更為重要.預防措施包括避免近親結婚,推行遺傳咨詢、攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生.早期診斷、早期治療、加強臨床護理,對改善患者的生活質量有重要意義.
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥治疗
肌張力障礙綜合征的西醫治療
(一)治療
1.病因治療如Wilson病驅銅治療及調整飲食可控制癥狀,藥物誘導肌張力障礙可停藥或換藥治療.
2.對癥治療藥物誘導急性肌張力障礙用苯海拉明(diphenhydramine)50mg或地西泮(安定)5~10mg緩慢靜脈註射,可快速緩解癥狀;也可口服地西泮(安定)10~20mg/d,苯海索(三己芬迪)6~30mg/d,L-dopa1~3g/d,氟哌啶醇(holoperidol)1.5~4.5mg/d,可使約50%的病例癥狀緩解.生活指導及生物反饋療法也有一定的治療價值.改善癥狀藥物種類雖較多,如地西泮(安定)、勞拉西泮(羅拉)、苯海索(安坦)、撲米酮(撲癇酮)、丙戊酸鈉(丙戊酸)、卡馬西平、巴氯芬(脊舒)、多巴胺受體激動藥、左旋多巴和鋰劑等,但無任何一種藥物對所有慢性肌張力障礙都有效,臨床應對不同患者通過反復療效觀察,尋找有效藥物和最佳治療劑量.
3.肉毒毒素A局限性肌張力障礙受累肌肉在肌電圖引導下註射肉毒毒素A有效率達95%,手部及頸部有效率85%,可維持3~4個月,反復註射療效維持時間縮短.
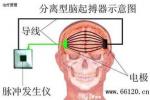
4.外科治療藥物及肉毒毒素A註射無效時可選擇手術,神經切斷術可解除血管對神經壓迫,廣泛性及偏側性肌張力障礙可選用丘腦損毀術.
(二)預後
不同類型預後不盡相同,一般為良性過程,如原發性書寫痙攣癥狀相當穩定,很少有擴散加重傾向.
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥饮食
宜吃芹菜、金針菜、韭菜、冬瓜、烏梅、柿餅、芝麻、蓮子、海參.
肌張力障礙綜合征 肌緊張不足綜合征 肌張力障礙 肌張力障礙綜合癥并发症
肌張力的變化常不引人註意,而異常的體位姿勢和不自主的變換動作更為引人註意.
2/2 首页 上一页 1 2


















