特發性炎癥性肌病
特發性炎癥性肌病百科
特發性肌炎-皮肌炎(polymyositis-dermatomyositis,PM-DM)是一組亞急性或慢性起病的獲得性炎癥性肌病,其主要病理特征是肌纖維壞死、再生及肌間質內炎性細胞浸潤,PM-DM的病因未明,到目前為止仍屬排除性診斷,凡是找不到明確感染因子(如病毒,細菌、寄生蟲等)的炎癥性肌病均屬此病范疇,故又稱為特發性炎癥性肌病(idiopathicinflammatorymyopathy).由於本組疾病對皮質類固醇激素治療反應良好,推測其發病機制可能與自身免疫異常有關.多發性肌炎(polymyositis,PM)的主要臨床表現是以肢體近端、頸部和咽部肌群無力伴疼痛為主的彌漫性炎性肌病.若合並典型皮疹者,則稱之為皮肌炎(dermatomyositis,DM).約1/3病人可合並其他結締組織疾病,1/10病人與腫瘤伴發.多發性肌炎-皮肌炎腎損害見於少數患者.基本知識醫保疾病:否患病比例:0.013%易感人群:無特殊人群傳染方式:無傳染性並發癥:無機堿皮膚損傷治療常識就診科室:免疫科內科治療方式:藥物治療支持性治療治療周期:7-14天治愈率:60%常用藥品:醋酸潑尼松片馬來酸氯苯那敏片治療費用:根據不同醫院,收費標準不一致,市三甲醫院約(1000--5000元)溫馨提示多食新鮮的水果和蔬菜,以保證維生素的攝入量.
特發性炎癥性肌病
特發性炎癥性肌病病因
病毒感染(30%)
PM和DM病因未明,可能為易感人群感染某些病毒後,機體免疫系統發生紊亂,導致以骨骼肌病變為主的結締組織炎癥,研究發現PM/DM與HLA-DR3,包涵體肌炎與HLA-DR1兒童DM與C4無效基因等高度相關,柯薩奇病毒,黏病毒,副黏病毒,埃可病毒,流感病毒,乙型肝炎病毒等感染可促發本病發作.
體液免疫異常(30%)
體液免疫異常表現為免疫球蛋白升高,自身抗體陽性,其中肌炎特異性抗體(致病性抗體)是直接針對肌細胞胞漿抗原成分的抗體,如抗Jo-1,抗Mi-2,抗PL-7抗體等,兒童DM皮膚活檢顯示小血管壁上有補體膜攻擊現象,提示皮膚肌肉損傷與補體活化有關.
細胞免疫異常(30%)
細胞免疫異常包括多種細胞因子數和量的異常,免疫病理證實壞死肌纖維周圍有CD8+T單核細胞浸潤,提示肌肉損傷由細胞免疫介導.

特發性炎癥性肌病
特發性炎癥性肌病症状
1.肌肉病變:表現為肌無力,肌痛,肌壓痛和肌萎縮,其中,以對稱性進行性肌無力最為突出,近端肢帶肌,頸肌和咽肌為常見受累肌群,表現為步行障礙,舉臂抬頭困難,嚴重者不能梳頭和穿衣,若動眼,咽,喉,食管,膈,肋間肌肉受累,可發生復視,斜視,聲嘶,吞咽困難,呼吸困難,心肌受累可發生心律失常和心力衰竭.
2.皮膚病變:皮損可與肌肉癥狀同時或較早或較晚出現.皮疹包括:
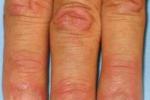
(1)Gottron征:掌指關節和近端指關節伸面紅色鱗狀斑丘疹,日久後中心萎縮,色素減退,本征為DM特異性皮疹,具診斷價值,發生率約為70%.
(2)向陽性皮疹(heliotroperash):眶周出現淡紅色水腫性斑疹,以上瞼為主,約50%早期即可出現此征,也為DM特征性皮疹之一.
(3)暴露部位皮疹:30%出現面,頸,胸部V字區,頸後披肩狀以及四肢暴露部位紅色皮疹,伴毛細血管擴張,部分對光敏感.
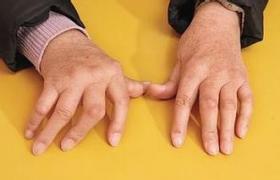
(4)技工手:1/3患者雙手外側和掌面皮膚出現角化,裂紋,脫屑,與職業性技工操作者的手相似.
3.其他表現:不規則低熱,可為初發癥狀或在病程中發生,20%伴關節病變,主要為關節痛,繼發於鄰近肌肉攣縮,可致關節畸形和活動受限,有20%~30%出現雷諾現象,少數頸部淋巴結可腫大,累及心臟者出現,心動過速,房顫,心肌損害,心臟擴大和心力衰竭.嚴重病例出現胸膜炎,間質性肺炎和肺功能下降,其中肺功能損傷常為主要致死原因,約1/3病例出現肝臟輕至中度腫大,消化道鋇餐可示食管蠕動差,擴張及梨狀窩鋇劑滯留,視網膜出現滲出,出血,脈絡膜炎等.
4.原發性多發性肌炎:約占炎性肌病病人的1/3,通常隱襲起病,在數周,數月,數年內緩慢進展,僅少數病人急性起病,在數日內出現嚴重肌無力,甚或橫紋肌溶解,此病可見於任何年齡,女性比男性多見,男女比例為1∶2.
(1)一般表現:病人可有畏寒,中度或低度發熱,疲乏,無力,食欲不振,體重減輕,少數病人可出現四肢關節痛,個別病人以關節炎為首發癥狀,並伴有晨僵,但關節腫脹一般不足6周,無關節畸形,需與類風濕關節炎鑒別,如病人手部出現畸形,一般為肌肉痙攣所致,無明顯關節破壞,少數病人可出現雷諾現象,表現為情緒激動或遇冷時出現指(趾)端皮膚蒼白,青紫,潮紅改變.
(2)肌肉表現:本病通常累及橫紋肌,病人首先感到四肢近端及頸部肌肉無力,一般兩側對稱,當病人有骨盆帶及下肢近端肌無力時,可表現為上樓梯,上坡困難,蹲下或從座椅上站起困難,步態蹣跚,走路時感下肢酸軟,當肩胛帶或上肢近端肌肉受累時,可出現抬臂困難,不能梳頭和穿衣,頸肌無力者平臥時抬頭困難,呼吸肌無力可造成胸悶,氣促,呼吸困難,嚴重者需借助呼吸機進行輔助呼吸,咽喉或上段食道橫紋肌受累可出現吞咽困難,攝入流質食物時經鼻孔流出,可引起嗆咳和誤吸,眼輪匝肌和面肌受累罕見,這有助於與重癥肌無力鑒別,對稱性近端肌無力為本病特點,但在整個病程中患者可出現不同程度的四肢遠端肌無力表現,體檢需確認個別肌肉或肌群有否無力,在每次隨診中應記錄肌無力的嚴重程度,肌力的系列定量估測對病人是一個重要的測量指標,因為實驗室指標不總能準確反映疾病活動性,已有幾個關於肌無力嚴重程度的分級方法,Rose及Walton的方法將體格檢查與肌肉功能綜合起來,簡便易行,另外,還有按年齡和性別標準快速評價下肢肌力的方法,一種改良的血壓計檢測方法可用來測量肩外展肌的肌力,簡便可重復,且可用於其他肌群的測量,用一種手持拉力計可測量多個肌群的肌力.
除肌無力外,25%病人可伴肌痛及(或)肌肉壓痛,少數病人僅有肌痛而無肌無力表現,對此類病人的診斷須高度謹慎,有時病人僅有乏力,需經仔細檢查方可發現其肌無力表現.
隨病程的延長,病人可出現不同程度的肌肉萎縮,早期病變肌肉質地可正常,出現纖維化改變後肌肉觸之變硬,罕見的暴發型病人表現為橫紋肌溶解,肌紅蛋白尿,腎功能衰竭.
(3)肺部表現:間質性肺炎,肺纖維化,胸膜炎是多發性肌炎最常見的肺部病變,可在病程中的任何時候出現,表現為胸悶,氣短,咳嗽,咯痰,呼吸困難,紫紺等,少數患者有少量胸腔積液,但單側大量胸腔積液少見,需註意與結核或腫瘤鑒別,由於食道運動障礙,吞咽困難,喉反射失調,常引起吸入性肺炎,肺不張等,如病人有呼吸肌無力,排痰困難,易導致細菌生長,由於免疫抑制劑的使用,常繼發細菌,黴菌和結核感染,所以肺部受累是多發性肌炎的常見死亡原因之一.
(4)心臟表現:50%的患者有心臟受累,主要為心肌炎和心包炎,心內膜炎和心肌梗死少見,病人可表現為心悸,氣短,胸悶,心前區不適,呼吸困難,病人可有心包積液,心臟擴大,心肌病,心律不齊,傳導阻滯等,晚期出現的充血性心力衰竭和嚴重心律失常是患者主要死亡原因之一.
(5)腎臟病變:病人可出現蛋白尿,血尿,管型尿,罕見的暴發型多發性肌炎可表現為橫紋肌溶解,肌紅蛋白尿,腎功能衰竭,腎組織活檢可有局部免疫球蛋白和補體沉積,為局灶性腎小球腎炎,提示免疫復合物可能是腎損害的原因.
5.原發性皮肌炎:除上述肌炎表現外,病人尚有特征性皮疹,55%的病人皮疹出現在肌炎之前,25%與肌炎同時出現,15%出現在肌炎之後.
(1)肌炎表現(見“原發性多發性肌炎”).
(2)皮膚表現:皮肌炎常見的皮膚表現有:
①向陽性皮疹(heliotroperash):為上眼瞼或眶周出現的水腫性暗紫紅色斑,可為一側或兩側,近瞼緣處可有毛細血管擴張,對光照較敏感,此種皮疹還可出現在兩頰部,鼻梁,頸部,前胸V形區和上背部,見於60%~80%的皮肌炎病人,這是皮肌炎的一種特征性皮疹.
②Gottron斑丘疹(Gottron’spapules):是一種米粒至綠豆大小的紅色或紫紅色斑丘疹,邊緣不整,可融合成片,伴有皮膚萎縮,毛細血管擴張和色素沉著或減退,偶有皮膚破潰,此類皮損出現於關節伸面,特別是掌指關節和指間關節伸面,亦可出現在肘,膝關節伸面及內踝等處,邊界清晰,表面覆有鱗屑或有局部水腫,可出現於60%~80%的皮肌炎病人,這是該病的又一特征性皮損.
③甲周病變:甲根皺襞處可見毛細血管擴張性紅斑,或出現瘀點,甲皺及甲床有不規則增厚,甲周可有線狀充血性紅斑,局部出現色素沉著或色素脫失.
④“技工手”樣變:在手指的掌面和側面出現污穢,深色的水平線橫過手指,因類似於長期用手工操作的勞動手,故名“技工手”.
⑤其他皮膚黏膜改變:20%的病人可有雷諾現象,由甲皺微循環改變所致,手指潰瘍,甲周梗死等皮膚血管炎表現亦可出現,且提示有惡性病變的潛在可能,口腔粘膜亦可出現紅斑,75%~80%的病人可出現光過敏,還可出現肌肉硬結,皮下小結,皮下鈣化改變.
6.惡性腫瘤相關的皮肌炎或多發性肌炎:在1935年,Ringel等首次報道瞭肌炎與惡性腫瘤相關,接下來的觀察報道提示多發性肌炎-皮肌炎病人患惡性腫瘤的危險性明顯增加,有人認為皮肌炎病人比多發性肌炎病人更易患腫瘤,雖然這組病人肌肉和皮膚改變與其他組病人無明顯差別,但已被獨立劃分出來,約占所有病例的10%(6%~60%),病人可先有惡性腫瘤,以後出現多發性肌炎或皮肌炎,也有的患者在患多發性肌炎或皮肌炎若幹年後發生惡性腫瘤,偶見兩種病變在1年內同時發生並有平行的病程,早期研究提示卵巢癌和胃癌最常見,其他腫瘤亦可出現,如肺癌,乳腺癌,消化道腫瘤,血液系統惡性腫瘤,甲狀腺癌,鼻咽癌,腎癌等.
一般在兒童肌炎和與結締組織病相關的肌炎病人腫瘤少見,40歲以上病人腫瘤發生率高,尤其是60歲以上老年病人,因此,對這類病人詳細詢問病史和全面體格檢查是非常重要的,特別是對乳腺,盆腔,直腸的檢查不容忽視,還可結合相應的輔助檢查,如血常規,生化,血蛋白電泳,癌胚抗原,免疫學檢查,尿紅細胞及細胞學分析,便潛血,胸片,痰細胞學檢查,骨掃描,B超等,以尋找有關腫瘤診斷的線索,必要時,可進行消化道造影,宮頸刮片等檢查,肌炎的病人出現惡性腫瘤的類型和部位與其性別和年齡有關.
有作者提出,肌炎是一種癌旁綜合征,其發病可能與機體免疫狀態的改變,腫瘤和肌肉間存在交叉反應抗原,或肌肉本身存在潛在的病毒感染有關.
7.兒童期皮肌炎或多發性肌炎:兒童期皮肌炎或多發性肌炎占肌炎病人的8%~20%,發病前常有上呼吸道感染史,無雷諾現象,很少有肺間質纖維化和惡性腫瘤,多發生在5~14歲,男女比例為1.3~2∶1,雖然偶有兒童與成人多發性肌炎病變過程相似的情況,但通常所觀察到的兒童炎性肌病過程有其獨特之處,兒童期皮肌炎的一般表現為皮疹和肌無力,但由於同時存在血管炎,異位鈣化和脂肪萎縮,使其與成人表現有很大區別.
(1)皮膚表現:通常患兒先出現皮膚表現,然後出現肌無力,皮疹一般較典型,是位於顴部和肘,手指,膝關節伸面的紅斑,可有脫屑,色素沉著和色素脫失,眶周亦可出現充血性丘疹,嚴重的急性期患者可出現皮膚血管炎表現,如皮膚潰瘍,甲周梗死,這些癥狀的出現提示可能有潛在的惡性病變.
(2)肌肉表現:肌無力,肌痛和僵硬在近端肌肉和頸部屈肌表現明顯,但也可為彌漫性,受累肌肉有壓痛和腫脹,在兒童期皮肌炎皮膚損害和肌無力幾乎同時出現,但這兩種表現的嚴重性和進展情況則有較大的個體差異,嚴重的肌無力可導致咀嚼困難,聲音嘶啞,吞咽和呼吸困難,偶可引起呼吸衰竭.
(3)血管炎:某些患兒,不經治療可完全緩解,但在伴有血管炎的嚴重病例,雖經治療亦不能阻止疾病進展,血管炎還可引起胃腸道潰瘍,出血或穿孔.
(4)異位鈣化:異位鈣化可出現在皮膚,皮下組織,肌肉或筋膜中,可為彌漫性或局限性,某些患兒皮下鈣化與血管炎同時出現,有些患兒則隻有皮下鈣化,鈣化處皮膚可出現潰瘍,影響患兒的姿勢,而且長期肌無力,肌肉攣縮,可影響活動能力.
(5)其他表現:部分病人可出現心包積液和胸腔積液,心電圖可出現傳導阻滯改變,急性期可出現視網膜水腫和出血,視神經纖維損傷,視神經萎縮,視野缺失或一過性視網膜剝離,個別病人可出現血小板減少,末梢神經炎,癲癇發作和蛛網膜下腔出血.
盡管兒童皮肌炎,多發性肌炎比成人預後好,但死亡人數仍達病人總數的1/3.
8.其他結締組織病相關的多發性肌炎或皮肌炎:約1/5的肌炎患者伴發其他結締組織病,形成重疊綜合征,這種重疊可能是由某種內在原因所致,而不象是隨機相疊,常見的與之重疊的疾病有系統性紅斑狼瘡,風濕性多肌痛,幹燥綜合征,類風濕關節炎,混合結締組織病,結節性多動脈炎,韋格納肉芽腫病,巨細胞動脈炎,過敏性肉芽腫,超敏性血管炎等,診斷依賴於兩種風濕病各自的診斷標準,有時,特發性炎性肌病的臨床表現可能成為這類病人的突出特點,特別是當肌炎與系統性硬化癥,系統性紅斑狼瘡,類風濕關節炎,混合結締組織病和幹燥綜合征並存時,而在血管炎綜合征則少見,此時肌無力常與動脈炎和神經受累有關,而與肌肉的非化膿性炎性改變無關.
另一方面,某些結締組織病病人經常出現肌無力和其他肌病的表現,如血清肌酸磷酸激酶水平增高和典型的肌電圖改變,使之不易與典型的多發性肌炎相區別,也有的病人,雖有肌無力,但不伴有肌酶水平的增高和肌電圖改變.
繼發於另一種彌漫性結締組織病的肌炎病人的肌肉組織學改變可能與多發性肌炎病人相同,但某些病人可有其不同的表現特點,如,硬皮病病人肌肉病變的特點是肌纖維大小不等,偶有單個肌纖維壞死,在肌束內和肌肉周圍可有結締組織增生,肌肉周圍血管有單個核細胞浸潤,系統性紅斑狼瘡病人肌肉組織學改變與成人皮肌炎相似,充血性炎性改變在類風濕關節炎罕見,在幹燥綜合征亦少見,常見2型纖維萎縮及非特異性改變或肌肉結構基本正常僅伴少量淋巴細胞浸潤,在嚴重的類風濕性血管炎病人偶可見到肌肉組織的動脈炎改變,混合性結締組織病病人的肌肉病理學可與皮肌炎或硬皮病相同.
某些病人肌無力可能與治療藥物的副反應有關,如糖皮質激素,D-青黴胺和抗瘧藥等,有的病人可能是由於細胞因子的作用,如白細胞介素-1,白細胞介素-6,腫瘤壞死因子等,需註意鑒別.
9.包涵體肌炎:由於這是一種少見病,很多醫生缺乏對其診斷的經驗,因此其確切患病率尚不清楚,有報道認為這類病人約占所有炎性肌病總數的15%~28%,一般散發,無傢族聚集傾向,兒童罕見,40歲以下少見,多發生在老年病人,常隱襲起病,進展緩慢,病程較長,有些病例在診斷前癥狀已存在瞭5~6年,其臨床表現與多發性肌炎有很多相同之處,其區別在於典型的多發性肌炎的特點是,肌無力可為局灶性的,遠端肌肉亦可受累,且常兩側不對稱,早期出現明顯的手指或前臂屈肌和小腿伸肌受累,往往具有特征性,肌痛和肌肉壓痛罕見,一般無皮疹,晚期20%的病人可出現吞咽困難,有時癥狀非常明顯,面肌無力罕見,尚無眼瞼下垂或眼肌麻痹的報道,心血管系統的表現與多發性肌炎相似.
隨肌無力的逐漸加重,可出現肌萎縮和深部腱反射減弱,有些病人,疾病可緩慢持續進展,有的病人疾病則靜止在某些肌肉的無力和萎縮,尚無包涵體肌炎與腫瘤並存的報道,但有時可合並以下疾病:間質性肺炎,硬皮病,系統性紅斑狼瘡,皮肌炎,幹燥綜合征,免疫性血小板減少癥,結節病,銀屑病,糖尿病等,這些疾病與包涵體肌炎共存的頻率並不高,而且其意義尚不清楚.
這種疾病通常對糖皮質激素和免疫抑制劑治療反應不佳,但某些病人經靜脈輸入免疫球蛋白後,病情可得到改善,這是一種慢性進展性疾病,發病5~10年後,病人可能會失去行走能力.
10.其他肌炎:
(1)嗜酸性粒細胞增多性肌炎:這是一類少見病,可能代表瞭嗜酸性粒細胞增多綜合征病譜的表現之一,其特點是亞急性發病,可有近端肌無力和肌痛,血清肌酶(特別是肌酸磷酸肌酶等)水平升高,肌電圖有肌病性改變,組織病理學除有肌炎性改變外,嗜酸性粒細胞浸潤是其特點,有的病人對糖皮質激素,甲氨蝶呤或白細胞置換治療反應尚好,該病包括幾種不同的亞類.
①嗜酸性粒細胞增多-肌痛綜合征(eosinophilia-myalgiasyndrome)見硬皮病.
②嗜酸性筋膜炎(eosinophilicfaciitis)見硬皮病.
③復發性嗜酸粒細胞增多性肌周炎(relapsingeosinophilicperimyositis)該病特點是頸部和下肢肌肉疼痛和壓痛,而無肌無力表現,常有血沉增快,外周血嗜酸性粒細胞增多,血清肌酸磷酸激酶有時增高,組織學檢查可見肌束膜有嗜酸性粒細胞浸潤,對糖皮質激素治療反應好.
(2)局灶性結節性肌炎:這是一種急性出現的綜合征,表現為局灶的炎性疼痛性結節,有時可依次出現在不同的肌肉,叫局灶結節性肌炎,病理表現和對治療的反應與多發性肌炎相似,當單個出現時,需註意與肌肉腫瘤(肉瘤或橫紋肌肉瘤)或增生性筋膜炎和肌炎鑒別,當多個出現時,需註意與結節性多動脈炎發生的肌肉梗死相鑒別.
特發性炎癥性肌病
特發性炎癥性肌病检查
輔助檢查:
1.血清CPK,LDH,GOT均升高,血清肌紅蛋白含量明顯增高,血清蛋白電泳α、r球蛋白及血清IgG、IgA、IgM增高.半數以上患者血沉快.
2.24小時尿中肌酸排出量可顯著增加,>1000mg/24小時,且與病情嚴重程度有關.
3.肌電圖:插入電位延長,可有肌強直樣放電活動,輕收縮時運動單位電位平均波幅降低,時限縮短,可有大量纖顫波,多相波增多,重收縮時出現低波幅幹擾相或病理幹擾相.
4.肌肉活檢:示變性,壞死,炎細胞浸潤,肌纖維腫脹,呈玻璃樣,顆粒性或空泡性變,間質水腫,血管周圍淋巴細胞和漿細胞浸潤.
5.心電圖:異常率可達40%左右,心動過速,心肌炎樣表現,或出現心律不齊等.
特發性炎癥性肌病预防
其發病機制可能與自身免疫異常有關,而自身免疫性疾病尚無有效的預防方法,防止感染,感冒以及寒冷或炎熱等誘發因素,是防治的重點;防治並發癥也是臨床醫療護理的重要內容.
特發性炎癥性肌病治疗
就診科室:免疫科內科
治療方式:藥物治療支持性治療
治療周期:7-14天
治愈率:60%
常用藥品:醋酸潑尼松片馬來酸氯苯那敏片
治療費用:根據不同醫院,收費標準不一致,市三甲醫院約(1000--5000元)
預防:
1.去除可能的誘因,如風寒,濕熱等不良因素對人體的侵襲.
2.加強身體鍛煉生活規律,註意勞逸結合.
3.加強營養,預防感染.
4.調暢情志,保持心情愉快.
多發性肌炎-皮肌炎中醫治療方法:
(1)毒熱型:治法:清營解毒,活血止疼.
方藥:用解毒清營湯合普濟消毒飲加減.生玳瑁10g雙花炭10g、連翹15g生地炭10g丹皮15g、赤芍15g、川連10g白茅根30g生薏米30g、赤苓皮10g元胡10g、川楝子10g.分析:生玳瑁、雙花炭、連翹清熱解毒;生地炭丹皮、白茅根、赤芍涼血活血;生薏米、赤苓皮清熱利濕消腫;元胡川楝子活血理氣止痛;川連清心火.毒熱重者加人工牛黃;高熱加羚羊角粉或犀角粉;關節痛重時加雞血藤、秦艽;氣虛者加黃芪、白術、茯苓;水腫重時加車前子、澤瀉.
(2)寒濕型:治法:溫經散寒,活血通絡方藥:溫經通絡湯加減.黃芪30g、黨參10g、白術10g、山藥15g茯苓10g、丹參10g、雞血藤30g、鬼箭羽15g、烏蛇6g秦艽10g、桂枝10g.分析:黃芪、黨參、白術、茯苓、山藥補中益氣健脾益腎;丹參雞血藤、鬼箭羽活血化瘀通絡;鳥蛇、秦艽祛風濕,解毒通絡;桂枝溫化寒濕.紅斑持久不退時加雞冠花、凌霄花.也可用獨活寄生湯加減.
(3)氣血兩虛型:治法:調和陰陽,補益氣血通絡.方藥:首烏藤15g、雞血藤15g、天仙藤15g、鉤藤10g、當歸10g、赤白芍10g、丹參15g、黃芪10g、黨參10g、白術10g、茯苓10g、熟地10g.分析:首烏藤、雞血藤、天仙藤、鉤藤調和陰陽;當歸赤白芍、丹參、熟地養血活血;黃芪黨參茯苓、白術健脾益氣.低燒時加地骨皮、銀柴胡沙參;關節疼時加秦艽;腹脹加枳殼、厚樸.
另外,還可用單方成藥.如:
①雷公藤多甙:用於激素效果不佳或減量中加用,60mg/d,3次/d口服.或昆明山海棠,每次3片,3次/d口服.
②亦可根據不同情況選擇使用中成藥如人參歸脾丸、當歸丸人參健脾丸、補中益氣丸、滋補肝腎丸、秦艽丸、全鹿丸或黃精丸、雞血藤片等.
治療原則:
1.盡早應用腎上腺皮質激素或免疫抑制劑;
2.對癥和支持治療,防治各種感染;
3.血漿交換療法;
4.大劑量丙種球蛋白治療;
5.頑固、重癥者全身放療.
用藥原則:
1.腎上腺皮質激素(地塞米松、潑尼松(強的松))、氫化可的松、甲基氫化潑尼松(甲基強的松龍))等對大多數多發性肌炎有效,為治療本病首選,一般主張早期以大劑量沖擊,中劑量鞏固治療,時間不少於3個月,小劑量維持時間不應短於2年.
2.對於伴有潰瘍病、高血壓和糖尿病,不能應用腎上腺皮質激素的病人,以及經正規激素治療3個月,肌無力和肌痛仍無改善者,均應改用或加用免疫抑制劑(環磷酰胺、硫唑嘌呤或甲氨喋呤),對合並惡性腫瘤者尤合適.
3.大劑量丙種球蛋白以及血漿交換治療對本病有治療作用,但需較高的醫療費用.
4.長期大量應用激素、免疫抑制劑等,要註意藥物副作用,加強對癥、支持治療,對合並感染者,應盡早使用足量有效抗生素.
一、應用皮質激素.強的松40-60mg/d頓服.病情穩定後逐步減量,病情危重者氫化考的松200-300mg/d,或地塞米松10-20mg/d加於10%葡萄糖內靜滴.病情穩定後改為口服,調節劑量至療效最好,副作用最小的程度為維持量,有時達2-3年之久,療程中,應間斷使用ACTH.
二、激素大劑量短療程無效者應考慮停藥,可換用免疫抑制劑,硫唑嘌呤每日100-200mg.註意副作用.苯丙酸諾龍25mg肌註2次/周,對減輕疼痛,緩解癥狀有效.
三、有吞咽困難者,應鼻飼,以保證足夠的營養.呼吸道阻塞或呼吸肌麻痹者須及時氣管切開及輔助呼吸.強力寧具有激素樣作用而無其副作用,可選用.40mgV.D1次/d.
四、有條件者可行血漿交換治療.
特發性炎癥性肌病饮食
1、多以清淡食物為主,註意飲食規律.
2、根據醫生的建議合理飲食.
3、該疾病對飲食並沒有太大的禁忌,合理飲食即可.
特發性炎癥性肌病并发症
多發性肌炎系一種以肌無力,肌痛為主要表現的自身免疫性疾病,主要累及肢帶肌,頸項肌,咽喉部肌群,如同時累及皮膚,稱皮肌炎,多見於20-40歲婦女,部分病人病前可有感染史,以對稱性肢體近端肌肉無力,疼痛和壓痛為主征,可累及咽肌,呼吸肌和頸肌,晚期可有肌萎縮,有的合並皮膚或內臟損害,或有惡性腫瘤.
1/2 1 2 下一页 尾页


















