苯丙酮尿癥 E70.101
苯丙酮尿癥 E70.101 百科
苯丙酮尿癥(phenylketonuria,PKU)是一種常見的氨基酸代謝病,是由於苯丙氨酸代謝途徑中的酶缺陷,使得苯丙氨酸不能轉變為酪氨酸,導致苯丙氨酸及其酮酸蓄積並從尿中大量排出.臨床主要表現為智能低下,驚厥發作和色素減少.本病屬常染色體隱性遺傳.其發病率隨種族而異,美國約為1/14000,日本1/60000,我國1/16500.
苯丙酮尿癥 E70.101
苯丙酮尿癥 E70.101 病因
[發病機制]
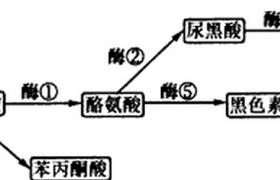
苯丙氨酸(phenylalanine,PA)是人體必需的氨基酸之一,正常小兒每日需要的攝人量約為200-500mg,其中1/3供合成蛋白,2/3則通過肝細胞中苯丙氨酸羥化酶(phenylalaninehydroxylase,PAH)的作用轉化為酪氨酸,以合成甲狀腺素、腎上腺素和黑色素等.苯丙氨酸
轉化為酪氨酸的過程中,除需PAH外,還必須有四氫生物蝶呤(tetrabiopterin,B11)作為輔酶參與.人體內的BH4是由鳥苷三磷酸(GTP),經過鳥苷三磷酸環化水合酶(GTP-CH)、6-丙酮酸四氫蝶呤合成酶(6-PTS)和二氫生物蝶呤還原酶(DHPR)等一系列酶的催化而合成.PAH、GTP-CH、DHPR三種酶的編碼基因分別定位於12q24.1、14q11,4p15.1-p16.1;而對6-PTS編碼基因的研究尚在進行中.上述任一編碼基因的突變都有可能造成相關酶的活性缺陷,致使苯丙氨酸發生異常累積.
註:PAH:苯丙氨酸羥化酶,BH4:四氫生物蝶呤(tetrabioptehn,),BH2:二氫生物蝶呤,GTP-CH:鳥苷三磷酸環化水合酶,6-PTS:6-丙酮酰四氫蝶呤合成酶,DHPR:二氫生物蝶呤還原酶
本病分為典型型和BH4缺乏型兩類:①典型PKU是由於患兒肝細胞缺乏PAH,不能將苯丙氨酸轉化為酪氨酸,因此苯丙氨酸在血、腦脊液、各種組織和尿液中的濃度極度增高,同時經旁路代謝產生大量的苯丙酮酸、苯乙酸、苯乳酸和對羥基苯乙酸,並從尿中排出.由於酪氨酸生成減少,致使甲狀腺素、腎上腺素和黑色素等合成不足,而蓄積的高濃度的苯丙氨酸及其旁路代謝產物導致細胞受損.②B1人缺乏型是由於GTP-CH、6-PTS或DHPR等任何一種酶缺乏所導致,BH4是苯丙氨酸、酪氨酸和色氨酸等芳香氨基酸在羥化過程中所必需的共同的輔酶,BH4的缺乏不僅苯丙氨酸不能轉變成酪氨酸,而且造成酪氨酸不能轉變成多巴胺,色氨酸不能轉變成5-羥色胺,多巴胺、5-羥色胺均為重要的神經遞質,其缺乏可加重神經系統的損害,故BH4缺乏型PKU的臨床癥狀更重,治療亦不易.
本病絕大多數為典型PKU,約1%左右為BH4缺乏型,其中約半數系6-PTS缺乏所致.

苯丙酮尿癥 E70.101
苯丙酮尿癥 E70.101 症状
[臨床表現]
出生時患兒正常,隨著進奶以後,一般在3-6個月時,即可出現癥狀,1歲時癥狀明顯.
1.神經系統 早期可有神經行為異常,如興奮不安、多動或嗜睡、萎靡;少數呈現肌張力增高、腱反射亢進,出現驚厥(約25%),繼之智能發育落後日漸明顯,80%有腦電圖異常.BH4缺乏型的神經系統癥狀出現較早且較嚴重,常見肌張力減 低、嗜睡、驚厥,如不經治療,常在幼兒期死亡.
2.外貌 因黑色素合成不足,在生後數月毛發、皮膚和虹膜色澤變淺.皮膚幹燥,有的常伴濕疹.
3.其他 由於尿和汗液中排出苯乙酸,呈特殊的鼠尿臭味.

苯丙酮尿癥 E70.101
苯丙酮尿癥 E70.101 检查
[診斷]
本病為少數可治性遺傳性代謝病之一,上述癥狀經飲食控制治療後可逆轉,但智能發後難以轉變,應力求早期診斷治療,以避免神經系統的不可逆損傷.由於患兒早期癥狀不典型,必須借助實驗室檢測.
1.新生兒期篩查 新生兒喂奶3日後,采集足跟末梢血,吸在厚濾紙上,晾幹後郵寄到篩查中心.采用Guthrie細菌生長抑制試驗半定量測定,其原理是苯丙氨酸能促進已被抑制的枯草桿菌重新生長,以生長圈的范圍測定血中苯丙氨酸的含量;亦可在苯丙氨酸脫氫酶的作用下進行比色定量測定,其假陰性率較低.當苯丙氨酸含量>0.24mmol/L(4mg/d1),即兩倍於正常參考值時,應復查或采靜脈血定量測定苯丙氨酸和酪氨酸.正常人苯丙氨酸濃度為0.06-0.18mmol/L(1-3mg/d1),而患兒血漿苯丙氨酸可高達1.2mmol/L(20mg/d1)以上,中酪氨酸正常或稍低.
2.尿三氯化鐵試驗 用於較大嬰兒和兒童的篩查.將三氯化鐵滴人尿液,如立即出現綠色反應,則為陽性,表明尿中苯丙氨酸濃度增高.此外,二硝基苯肼試驗也可以測尿中苯丙氨酸,黃色沉淀為陽性.
3.血漿氨基酸分析和尿液有機酸分析 可為本病提供生化診斷依據,同時也可鑒別其他的氨基酸、有機酸代謝病.
4.尿蝶呤分析 應用高壓液相層析(HPLC)測定尿液中新蝶呤和生物蝶呤的含量,鑒別各型PKU.典型PKU患兒尿中蝶吟總排出量增高,新蝶呤與生物蝶呤比值正常;DHPR缺乏的患兒蝶呤總排出量增加,四氫生物蝶呤減少;6-PTS缺乏的患兒則新蝶吟排出量增加,其與生物蝶呤的比值增高;GTP-CH缺乏的患兒其蝶吟總排出量減少.
5.酶學診斷 PAH僅存在於肝細胞,需經肝活檢測定,不適用於臨床診斷.其他3種酶的活性可采用外周血中紅、白細胞或皮膚成纖維細胞測定.
6.DNA分析 該技術近年來廣泛用於PKU診斷、雜合子檢出和產前診斷.但由於基因的多態性,分析結果務須謹慎.
苯丙酮尿癥 E70.101 预防
苯丙酮尿癥 E70.101 治疗
[治療]
診斷一旦明確,應盡早給予積極治療,主要是飲食療法.開始治療的年齡愈小,效果愈好.
1.低苯丙氨酸飲食 主要適用於典型PKU以及血苯丙氨酸持續高於1.22mmol/L(20mg/d1)的患者.由於苯丙氨酸是合成蛋白質的必需氨基酸,完全缺乏時亦可導致神經系統損害,因此對嬰兒可喂給特制的低苯丙氨酸奶粉,到幼兒期添加輔食時應以淀粉類、蔬菜、水果等低蛋白食物為主.苯丙氨酸需要量,2個月以內約需50~70mg/(kg·d),3-6個月約40mg/(kg·d),2歲約為25~30mg/(kg·d),4歲以上約10~30mg/(kg·d),以能維持血中苯丙氨酸濃度在0.12~0.6mmol/L(2~10mg/d1)為宜.飲食控制至少需持續到青春期以後.
2.BH4、5-羥色胺和L-DOPA 主要用於Bfl缺乏型PKU,除飲食控制外,需給予此類藥物.
苯丙酮尿癥 E70.101 饮食
飲食原則是:既要限制飲食中苯丙氨酸攝入量,又要給予足量的生長發育所需要的營養物質.飲食治療如果在出生後5個月才開始,則大部分患兒有智力低下,4~5歲時才開始飲食治療,則隻能使抽搐發作和行為異常得到減輕.新生兒隨著年齡的增大,苯丙氨酸用於蛋白質合成的量將逐漸減少,需要代謝清除的量則逐漸增多,如果有PAH活性缺乏,苯丙酮酸也日益增多,因此飲食中的苯丙氨酸的量應隨年齡增長而逐漸減少.
苯丙酮尿癥 E70.101 并发症
約2/3的患兒有輕度小顱畸形,眼底正常,無內臟腫大或骨骼異常.
1/2 1 2 下一页 尾页


















