小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病百科
本病為神經皮膚綜合征(neurocutaneoussyndrome)中最常見的一種綜合征,是一些起源於外胚層的組織和器官發育異常的疾病.常表現為神經、皮膚和眼睛的異常,有時也波及中胚層或內胚層發育的器官.由於受累的器官、系統不同,臨床表現多種多樣.目前已知此類疾病多達40餘種,但常見的隻有3種,即神經纖維瘤病,結節性硬化癥及腦面部血管瘤病(Sturge-Weber綜合征),這些疾病多表現為常染色體顯性遺傳,有一個較高的、不完全的外顯率.目前對這類疾病的病因瞭解得還不清楚,可能與胚胎發育早期出現某些變異有關.
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病病因
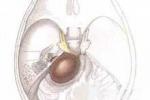
(一)發病原因為常染色體顯性遺傳,外顯率變異性較大,目前已知本病在染色體上的基因定位,Ⅰ型在17染色體上(17q11);Ⅱ型在22染色體(22q11→13),多發性神經纖維瘤病為胚胎形成早期階段神經嵴分化和遷徙異常導致的多系統損害的,常見的常染色體顯性遺傳病.(二)發病機制本病變化多端,各個系統及器官均可受累,出生即有,呈慢性進展特征,並發癥多出現於10年後,目前根據臨床表現,細胞生物學和分子生物學特點將其分為Ⅰ型神經纖維瘤病(neurofibromatosistypeⅠ,NFⅠ)亦稱vonRecklinghausen病和Ⅱ型神經纖維瘤病(neurofibromatosistypeⅡ,NFⅡ),即雙側聽神經瘤.1.Ⅰ型神經纖維瘤病:主要病理特征為沿著粗大的末梢神經生長的腫瘤(即分佈於脊神經,腦神經,皮膚或皮下神經的多發性神經纖維瘤及神經鞘瘤),腫瘤大小不一,與神經鞘膜緊密聯結,附有中胚層的神經束膜和外膜的細胞,皮膚或皮下神經纖維瘤大多數位於真皮和侵入皮下組織,病變界線不清,腫瘤無包膜,內有成熟的新生膠原纖維,無髓鞘及有髓鞘的纖維軸索摻雜,並可見到成團的施萬細胞,中樞神經系統亦可見腫瘤發生,最常見的是視神經膠質瘤,在基底節和丘腦部位也可出現膠質瘤,在Ⅱ型中為聽神經瘤及腦膜瘤,也可出現其他腫瘤,如Wilms瘤,成神經細胞瘤和嗜鉻細胞瘤等,一般來說不論中樞神經或是末梢神經的神經纖維瘤多為良性,但也可惡性變,大約有5%轉變為神經纖維肉瘤.2.Ⅱ型神經纖維瘤病:聽神經瘤病理改變屬前庭神經鞘瘤,NFⅡ常合並有腦膜瘤,脊膜瘤,星形細胞瘤以及脊旁後根神經鞘瘤,皮膚腫瘤以神經鞘瘤為主,偶有皮膚神經纖維瘤,極少出現叢狀神經纖維瘤.
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病症状
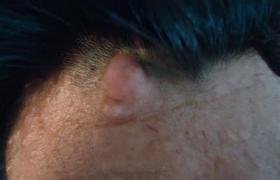
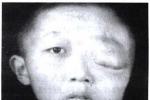
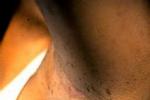
1.皮膚改變:咖啡牛奶斑是本病的重要體征,出生時即可發現,為一些淺棕色(咖啡裡加牛奶的顏色)斑,大小不等,形狀不一,不隆起於皮膚,不脫屑,感覺無異常,除手掌,足底和頭皮外,軀體其他部位均可波及,正常小兒有時也可見到1~2塊咖啡牛奶斑,無診斷意義,6塊以上直徑大於5mm的咖啡牛奶斑則有診斷價值,有時在腋窩鼠蹊部或軀幹其他部位見到一些1~3mm直徑似雀斑狀的淺棕色斑,稱為腋窩雀斑,成簇出現,數目往往較多,也具有診斷意義.皮膚的神經纖維瘤在嬰幼兒時期往往不明顯,青春期後增多,表現為結節狀隆起,有時有蒂,與皮膚色澤一致或呈暗紅色,大小由數毫米至數厘米,數目多少不等,多見於軀幹,四肢及頭部較少,如腫瘤壓迫神經可引起疼痛或功能障礙,叢狀神經纖維瘤常波及面部,兒童期也可見到,常破壞面容,頸部或縱隔的叢狀神經纖維瘤可壓迫呼吸道,影響呼吸.2.眼部異常:虹膜部位常可見到色素性虹膜錯構瘤(Lisch小體),一般體格檢查不能發現,需在裂隙燈下觀察,表現為突起的褐色斑塊,邊緣清晰,無特殊癥狀,6歲以後常見,有診斷意義.3.神經系統:神經纖維瘤組織在病理上屬錯構瘤樣結構,為良性腫瘤,身體各部位的神經纖維均可受累,由於腫瘤的性質,部位的不同臨床表現也多種多樣,視神經膠質瘤可見於15%的病人中,表現為進行性視力喪失,視神經萎縮,局部疼痛或眼球突出,單側或雙側,聽神經瘤往往在10歲以後發生,表現為聽力喪失,耳鳴,眩暈及面肌無力,腦部還可見到腦膜瘤,星形細胞瘤及室管膜瘤,脊髓及神經根也可波及,本病可伴有學習困難及行為障礙,但明顯智力低下及癲癇發作不太多見.4.其他系統:骨骼常表現為先天性骨發育不良,骨皮質變薄,鈣化不全等,常可見蝶骨發育不良,病理性骨折,脛骨假關節形成等.1.Ⅰ型神經纖維瘤病具有下列2項或2項以上:(1)6個或6個以上咖啡牛奶斑,青春期前直徑大於5mm,青春期後15mm.(2)腋窩雀斑.(3)視神經膠質瘤.(4)2個以上神經纖維瘤或1個叢狀神經纖維瘤.(5)一級親屬中有Ⅰ型神經纖維瘤.(6)2個或更多的Lisch小體.(7)骨病變.2.Ⅱ型神經纖維瘤病(1)雙側聽神經瘤(需經MRI,CT或組織學證實).(2)一側聽神經瘤,同時一級親屬中有Ⅱ型神經纖維瘤病.(3)一級親屬中有Ⅱ型神經纖維瘤病,且患者本人有下列任何兩種疾病:神經纖維瘤,腦(脊)膜瘤,神經鞘瘤,神經膠質瘤.
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病检查
目前已應用一種直接基因診斷技術-蛋白截斷分析(proteintruncationassay),結合基因連鎖和突變分析可定出很多NFⅠ基因的突變株,使對NFⅠ的基因診斷和產前診斷成為可能,NFⅠ基因定位於染色體17q11.2,隨著突變檢測技術的完善,即基因擴增和等位基因特異性寡核苷酸雜交法,限制性長度片段多態性連鎖分析,蛋白截斷分析法,錯配化學斷裂法及變性梯度凝膠電泳法的綜合應用可提高NFⅠ產前診斷和癥狀前診斷的準確率和可靠性,NFⅡ的多數患者是突變的結果.必要時可做皮膚和皮下結節或神經幹包塊的病理活檢.1.基礎檢查:臨床考慮本病患兒應作如下基礎檢查,如電測聽,腦幹誘發電位,視覺誘發電位,腦電圖,心理測驗(包括預測學習能力)等,可發現腦幹聽覺誘發電位等各種異常.2.X射線照片:頭顱內聽道X射線片示雙側內聽道破壞;骨骼X射線檢查有助於發現骨骼畸形,該病骨骼X射線片上可見大的,多發的囊狀結構,可見透明區,骨皮層變薄.3.椎管造影:有助於發現中樞神經系統腫瘤.4.MRI成像:頭顱MRI成像示雙側聽神經瘤,在蒼白球,丘腦和內囊部位顯示不正常的信號,這可能意味著存在低惡性度的神經膠質瘤或錯構瘤,而CT掃描則檢測不到,這些可以是學習困難,註意力不集中和語言障礙的原因,MRI示大腦白質斑塊狀異常信號,TW1低信號,TW2高信號,性質不明,暫稱之為不明原因發光物質,無癥狀病人應每年反復查體,進行神經學評價,包括血壓,聽覺和視覺的篩查,探查神經纖維瘤病的並發癥.5.腦或視神經CT掃描:CT可發現中樞神經系統腫瘤,但約85%患者顱腦CT無異常,腦室大小正常的巨頭很常見,繼發於導水管狹窄的腦水腫則少見.
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病预防
通過對某一改變的DNA序列進行特征性單鏈構象多態性分析,為檢查胎兒DNA提供瞭精確的產前檢查方法,在傢族病例中(有受累及未受累的傢族成員)利用連鎖分析加之產前診斷,能達到一定精確程度,必要時可終止妊娠.
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病治疗
(一)治療當腫瘤壓迫神經系統有臨床癥狀時,可行手術切除,放射治療無效.1.Ⅰ型神經纖維瘤病無特殊治療,當腫瘤生長迅速且有劇痛時應切除以防惡變,對叢狀神經纖維瘤和中樞型神經纖維瘤,應盡早施行手術,蔓狀神經纖維瘤病變部位多為小蔓狀血管竇樣改變,血管極為豐富,因此手術時應註意在病變外正常組織處切口,徹底切除腫瘤及其周圍組織,術後還可利用激光技術“清掃”,以防復發,單側眶板缺如者可予修補,癲癇發作者應予抗癇治療.2.Ⅱ型神經纖維瘤病本病主要是對癥治療,手術治療效果差,容易復發,因此原則上這類病人不適於做手術治療,較小的腫瘤可采用聚集放射療法,深部X射線及鐳放射治療無效,而腫瘤大到威脅病人生命時應考慮手術.(二)預後目前尚無有效的治療措施能阻止或逆轉NFⅠ的病程;Ⅱ型神經纖維瘤病癥手術治療效果差,容易復發,因此,防治關註的焦點集中在基因診斷和基因治療.
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病饮食
註意平時的生活習慣,註意多吃一些營養含量高的食物.
小兒神經纖維瘤病 小兒雷克林霍曾氏病 小兒雷克林霍曾病 小兒多發性神經纖維瘤病并发症
神經纖維瘤可能阻塞腦血管致使偏癱和智力障礙,由於本病的嚴重性和不確定性,所以存在心理障礙是不足為怪的,NFⅠ病人中惡性腫瘤也是值得註意的問題,神經纖維瘤偶爾分化成神經纖維肉瘤或惡性神經鞘瘤;嗜鉻細胞瘤,橫紋肌肉瘤,白血病和Wilms病的發病率比一般人群高,然而,在NFⅠ患者中中樞神經系統腫瘤(包括視神經膠質瘤,腦和脊髓的腦脊膜瘤,神經纖維瘤,星形細胞瘤和神經鞘瘤)發生率很高,故其是患者發病和死亡的重要原因.腫瘤壓迫神經可引起功能障礙,壓迫呼吸道影響呼吸,發生進行性視力喪失或眼球突出,聽力喪失,耳鳴,眩暈,伴有學習困難及行為障礙,常可見病理性骨折,脛骨假關節形成等,少數可伴發驚厥,語言和運動發育遲緩.
1/2 1 2 下一页 尾页



















