遺傳性腎炎 Alport綜合征 兒童腎積水
遺傳性腎炎 Alport綜合征 兒童腎積水百科
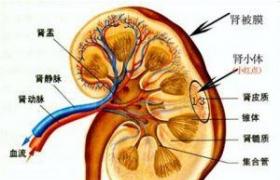
遺傳性腎炎(即Alport綜合征,AS)是一種主要表現為血尿、腎功能進行性減退、感音神經性耳聾和眼部異常的遺傳性腎小球基底膜疾病,是由於編碼腎小球基底膜的主要膠原成分-IV膠原基因突變而產生的疾病.基因突變的發生率約為1/10000~1/5000.根據遺傳方式可分為①X連鎖顯性遺傳(X-linkeddominant,XL,約占80%),致病基因在X染色體上,遺傳與性別有關.②常染色體隱性遺傳(autosomalrecessive,AR;約占15%),致病基因在常染色體上.③常染色體顯性遺傳(autosomaldominant,AD;極少數).分別因編碼IV型膠原不同ɑ鏈的基因COL4A5和(或)COL4A6、COL4A3和(或)COL4A4突變所致.
遺傳性腎炎 Alport綜合征 兒童腎積水
遺傳性腎炎 Alport綜合征 兒童腎積水病因
IV型膠原6條ɑ鏈聚合成3條三股螺旋分子結構稱為單體,單體聚合形成二聚體或四聚體,再相互絞連形成膠原網狀結構.X連鎖顯性遺傳Alport綜合征得基因突變主要發生在編碼IV型膠原ɑ5鏈的基因(COL4A5).常染色體隱性遺傳Alport綜合征的基因突變則發生在2號染色體上編碼IV型膠原ɑ3鏈或者ɑ4鏈的基因(COL4A3/COL4A4)上.
遺傳性腎炎 Alport綜合征 兒童腎積水
遺傳性腎炎 Alport綜合征 兒童腎積水症状
腎臟表現
以血尿最常見,大多為腎小球性血尿,來自我國的資料曾報告68%的Alport綜合征患者為腎小球性血尿.X連鎖顯性遺傳型的男性患者表現為持續性鏡下血尿,甚至可在出生後幾天內出現血尿;鏡下血尿的外顯率為100%.約67%的Alport綜合征男性患者有發作性肉眼血尿,多數在10~15歲前,肉眼血尿可在上呼吸道感染或勞累後出現.有作者認為X連鎖顯性遺傳型Alport綜合征傢系中的男孩,如果至10歲尚未發現血尿,則該男孩很可能未受累.X連鎖顯性遺傳型Alport綜合征的女性患者90%以上有鏡下血尿,少數女性患者出現肉眼血尿.幾乎所有常染色體陰性遺傳型的患者(無論男女)均表現血尿;而常染色體隱性遺傳型的雜合子親屬中,血尿發生率為50~60%,不超過80%.
X連鎖顯性遺傳型Alport綜合征男性患者均會出現蛋白尿,隨年齡增長或血尿出現而表現為持續蛋白尿,甚至出現腎病范圍蛋白尿,腎病綜合征的發生率為30~40%.國內北京大學第一醫院報道蛋白尿達腎病綜合征水平者占31.8%,並提示預後不佳.同樣,高血壓的發生率和嚴重性也隨年齡而增加,且多發生於男性患者.
聽力障礙
Alport綜合征患者聽力障礙表現為感音神經性耳聾,發生於耳蝸部位.耳聾為進行性,雙側不完全對稱,初為高頻區聽力下降,須借助聽力計診斷,漸及全音域,甚至影響日常的對話交流.目前,尚無先天性耳聾的報道.X連鎖顯性遺傳型Alport綜合征男性發生耳聾的幾率高於女性,發生年齡也較女性早.有報道X連鎖顯性遺傳型Alport綜合征男、女耳聾的發生率分別為81%和19%.而常染色體隱性遺傳型Alport綜合征約66.6%的患者於20歲前即表現出感音神經性耳聾.
眼部病變
Alport綜合征特征性眼部病變包括前園錐形晶狀體、眼底黃斑周圍點狀和斑點狀視網膜病變以及視網膜赤道部視網膜病變.前圓錐形晶狀體表現為晶狀體中央部位突向前囊,患者可表現為進行性近視,甚至導致前極性白內障或前囊自發穿孔.前圓錐形晶狀體多於20-30歲時出現,迄今報道的最小患者為13歲男性,有60-70%的X連鎖型男性、10%X連鎖顯性遺傳型女性以及約70%的常染色隱性遺傳型Alport綜合征患者出現前圓錐形晶狀體.Alport綜合征特異性的視網膜病變通常不影響視力,用眼底鏡或視網膜攝像的方法可見眼底黃斑周圍或視網膜赤道部有暗淡、甚至蒼白的點狀和斑點狀病灶,病變會伴隨腎功能的減退而進展.約70%X連鎖顯性遺傳型男性、10%的X連鎖顯性遺傳型女性以及約70%的常染色隱性遺傳型Alport綜合征患者出現視網膜病變且常與耳聾和前圓錐形晶狀體並存,但視網膜病變出現時間早於前錐形晶狀體.目前,尚未見常染色體顯性遺傳型Alport綜合征患者伴眼部受累的報道.
血液系統異常
目前認為AMME綜合征是伴有血液系統異常的Alport綜合征,主要表現為Alport、精神異常遲緩、面中部發育不良以及橢圓形紅細胞增多癥.研究證實此類Alport綜合征COL4A5基因全部缺失,且基因缺失范圍超越3‘端.此外,以往報道的血液系統異常,如巨血小板、血小板異常伴白細胞包涵體以及僅有血小板異常等表現並伴有“Alport樣”表現的疾病,已證實是編碼非肌球蛋白重鏈9的基因MYH9突變引起,而不是IV型膠原基因的突變所致.因此,此類疾病並非Alport綜合征,稱為MYH11A綜合征,為常染色體顯性遺傳.
彌漫性平滑肌瘤
某些青少年型Alport綜合征傢系或患者伴有顯著的平滑肌肥大,食管、氣管和女性生殖道(如陰蒂、大陰唇及子宮等)為最常見受累部位,並出現相應癥狀,如吞咽困難和呼吸困難等.
其他
有作者報道瞭某些病變,如甲狀腺疾病、IgA缺乏癥、腦橋後神經炎、升主動脈動脈瘤、肛門直腸畸形、精神病、纖維肌結構不良、I型神經纖維瘤病及Turner樣綜合征等.目前,上述病變尚不能確定為Alport綜合征特異性的臨床表現,很可能僅為Alport綜合征共存的疾病.
遺傳性腎炎 Alport綜合征 兒童腎積水
遺傳性腎炎 Alport綜合征 兒童腎積水检查
血尿和蛋白尿,男性患者表現為持續性鏡下血尿.開始時隻是微量蛋白尿,尿蛋白隨著年齡的增長逐漸增加,常發展至腎病綜合征蛋白尿.可有血小板缺陷及明顯出血傾向.發生腎功能衰竭時可有尿素氮、肌酐增高等改變.
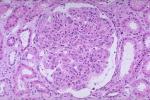
1.光鏡光學顯微鏡下腎臟病變無特異性.疾病早期腎小球病變大致正常,僅有輕度局灶節段性系膜組織增生,隨病變進展,腎小球漸發展至腎小球硬化,晚期腎小球出現纖維化及球性硬化,腎間質可從炎癥細胞浸潤發展到纖維化,並伴腎小管萎縮.
本病在腎臟皮、髓質交界處常見間質泡沫細胞.此泡沫細胞胞質含有中性脂肪、黏多糖、膽固醇及磷脂.該病變非本病特異,但在本病出現率高,對提示本綜合征仍有重要意義.
另外,有10%~25%的Alport綜合征病人具有胎兒型腎小球.胎兒型腎小球也能見於非Alport綜合征兒童,尤其是先天性腎病綜合征嬰兒,但5歲後,非Alport綜合征就很難再看到這一病變.此胎兒型腎小球主要見於10歲前患兒,尤其是5歲前的嬰幼兒.成人Alport綜合征患者少見.
2.電鏡腎小球基底膜(GBM)的超微結構改變對本病有診斷意義,而且早於光學顯微鏡改變.其主要病變有3種:GBM增厚、變薄及兩者相間.變薄的GBM常僅達正常厚度的1/4,多見於兒童及女性;增厚的GBM的可達正常厚度的2~5倍,其上皮側緣常呈不規則波浪形,增厚的致密帶縱向劈裂分層,相互交錯成網,網眼中含有類脂顆粒,多見於成人及男性.如果增厚的GBM廣泛存在,並與變薄的GBM相間出現,對本病診斷極有意義.不伴GBM增厚的純GBM變薄更常見於良性傢族性血尿即薄基底膜性腎病.有作者發現GBM增厚及破裂程度與蛋白尿程度相平行,GBM明顯增厚及破裂者疾病常進展,預後差(男性患者尤其如此).
3.免疫熒光免疫熒光和免疫組織化學多數呈陰性,提示無體液免疫參與致病.但偶爾也能見到少數腎小球毛細血管有IgM和C3沉積.免疫熒光檢查還發現,Alport綜合征病人的GBM缺乏Goodpasture抗原,也缺乏淀粉樣蛋白P.淀粉樣蛋白P存在於正常人的血漿和GBM中,Alport綜合征患者GBM缺乏淀粉樣蛋白P的意義尚待研究.
又有作者用Alport綜合征患者腎移植後產生的抗GBM抗體或直接用抗α5(Ⅳ)抗體,去孵育酸-尿素處理的本病患者皮膚切片,結果Alport綜合征男性患者的表皮基底膜完全不著色,女性患者僅節段性著色,與GBM染色結果相似,該發現在理論上提示本病患者的GBM及表皮基底膜缺乏Goodpasture綜合征抗原,而在實踐上有可能提供一個診斷本病的手段.
遺傳性腎炎 Alport綜合征 兒童腎積水预防
應註意避免感染、勞累及妊娠,還應禁用腎毒性藥物以預防本病發生.
遺傳性腎炎 Alport綜合征 兒童腎積水治疗
藥物幹預
近年來有報道環孢素和血管緊張素轉化酶抑制劑對於減少Alport綜合征患者尿蛋白、延緩腎臟病變發展至終末期腎病的進程方面均有積極的作用.醛固酮受體阻斷劑及血管緊張素受體拮抗劑可以減少Alport綜合征患者的尿蛋白.然而這些研究因缺少充分的循證醫學證據,其療效尚無定論.
腎臟替代治療
對於Alport綜合征進展至終末期腎病患者,可行透析或腎移植治療.腎移植是該病有效的治療措施,但國外報道約3~5%的接受腎移植的Alport綜合征患者移植後體內對被移植腎的正常腎小球基膜產生抗體,進而發生抗腎小球基底膜腎炎,致使移植失敗.此外,還有作者報道因移植後發生抗腎小球基膜腎炎、移植失敗者再移植仍可再次發生抗腎小球基底膜腎炎.
關於Alport綜合征腎移植供體選擇多認為以選擇活體腎移植為好,但若已發生移植腎後抗基底膜腎炎,在移植最後不用活體腎臟.有研究認為,雜合COL4A5基因女性攜帶者,臨床表現沒有蛋白尿、高血壓、腎功能減退和耳聾,可以作為供腎者,但移植後發生腎功能不全的幾率會高於移植健康供體的腎臟.
基因治療
盡管近年來已確定瞭各種遺傳型Alport綜合征的突變基因,且對Alport綜合征動物模型的基因治療取得瞭一定的結果,但目前基因治療仍存在一系列問題,如基因轉染效率不高、靶基因的導入途徑、導入時間時機的選擇、體內生存時間、病毒等載體的安全性及靶基因導入後調控等問題都未能很好解決,因此Alport綜合征的基因治療用於臨床尚需待以時日.
遺傳性腎炎 Alport綜合征 兒童腎積水饮食
1、避免過度勞累.
2、飲食宜清淡,禁忌酸辣刺激性食物.
遺傳性腎炎 Alport綜合征 兒童腎積水并发症
傳性腎炎傢族中,患者常並發許多非特異性病變,如甲狀腺疾病、IgA缺乏、球後視神經炎、升主動脈瘤、肛門直腸畸形、精神病和纖維肌性發育不良等.
1/2 1 2 下一页 尾页


















