小兒肝豆狀核變性
小兒肝豆狀核變性百科
肝豆狀核變性(hepatolenticulardegeneration,HLD)又稱Wilson病,是一種遺傳性銅代謝缺陷病,屬常染色體隱性遺傳.其特點是由於銅沉積在肝、腦、腎和角膜等組織,而引起一系列臨床癥狀.發病率約為1/(50萬~100萬).
小兒肝豆狀核變性
小兒肝豆狀核變性病因
本病致病基因定位在13q14.3.其發病機制迄今未明,現認為其基本代謝缺陷是肝臟不能正常合成血漿銅藍蛋白(ceruloplasmin),銅與銅藍蛋白的結合力下降以致自膽汁中排出銅量減少.人銅藍蛋白基因位於3q23-25,其基因突變與本病相關,目前發現6種移碼突變導致編碼蛋白功能障礙,銅藍蛋白無法與銅結合.
銅是人體所必需的微量元素之一.人體新陳代謝所需的許多重要的酶,如過氧化物歧化酶、細胞色素C氧化酶、酪氨酸酶、賴氨酸氧化酶和銅藍蛋白等,都需銅離子的參與合成.但機體內銅含量過多,高濃度的銅會使細胞受損和壞死,導致臟器功能損傷.其細胞毒性可能是銅與蛋白質、核酸過多結合,或使各種膜的脂質氧化,或是產生瞭過多的氧自由基,破壞細胞線粒體、過氧化物小體、溶酶體等.

小兒肝豆狀核變性
小兒肝豆狀核變性症状
小兒肝豆狀核變性
小兒肝豆狀核變性检查
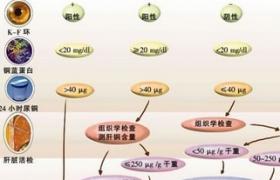
主要是血清銅藍蛋白降低,血清中非銅藍蛋白的銅增多,尿銅排出量增加,肝含銅量增加.
1.血清銅藍蛋白測定 正常小兒為200-400mg/L(或血清氧化酶測定為0.25~0.49O.D);患兒通常低於200mg/L(或<0.25O.D)甚至在50mg/L以下.但有5%的患兒正常或在正常低限.
2.24小時尿銅排出量測定 正常小兒尿銅低於40ug/24小時;患兒明顯增高,常達100-1000ug/24小時.由於其他原因所致肝病,包括慢性活動性肝炎、膽汁滯留、肝硬化等,亦常有尿銅排出量增高,在診斷時應予以鑒別.該項指標對估價治療效果和指導藥物劑量頗有幫助.
3.肝細胞含銅量測定 上述銅生化測定未能確診的病例,可采用肝穿刺方法測定肝組織內的銅含量.正常人肝含銅量多在20ug/g(幹重)以下,患兒可高達200~3000ug/g(幹重).采集肝標本時須註意勿被污染,送檢標本量應>5mg,以保證檢測數據可靠.肝銅量增高還可見於肝內、外膽管阻塞性膽汁瀦留、膽汁性肝硬化,應予以區別.
4.同位素銅結合試驗 根據正常人在經靜脈給銅後肝細胞能迅速將其合成銅藍蛋白並分泌入血循環的特點,可一次給患者靜註64Cu或67Cu(半衰期分別為12和61小時)0.3~0.5uCi.在註射後5~10分鐘、1、2、4、24和48小時各采集血樣一次,檢測其放射量.正常人在4~48小時之間呈持續上升,而患者則在4小時以後持續下降,其48小時血樣的計數僅為小時的一半.
小兒肝豆狀核變性预防
是否為缺陷基因攜帶者可檢出本癥雜合子,以便做遺傳咨詢;本病產前診斷已有可能,必要時可終止妊娠.
小兒肝豆狀核變性治疗
本病是可治性的,治療愈早,預後愈好.治療的原則是減少銅的攝入和增加銅的排出,避免銅在體內的沉積,以恢復和改善正常功能.
1.低銅飲食 每日食物中含銅量不應>lmg,不宜進食動物內臟、魚蝦海鮮、堅果、巧克力和蘑菇等含銅量高的食品.
2.促進銅排出 D-青黴胺(D-penicillamine)是目前最常用的藥物,能與銅離子絡合,促進尿銅排出,且可能促進細胞合成金屬硫因.劑量為每日20mg/kg,分次口服.治療期間應監測尿銅,第1年內要求每日尿銅排出量>2mg.一般在治療數周後神經系統癥狀可改善,而肝功能好轉常需3~4個月的治療,可根據尿銅及臨床癥狀調整用藥.因青黴胺可能拮抗維生素B6,故應每日補充維生素B625mg.青黴胺的副作用為藥物疹、血小板減少、腎病、關節炎等,其發生率不高.若不能使用,可考慮用鹽酸三乙撐四胺(triethylene-tetraminedihy-drochlorate),劑量為每日0.5-2g.近年來應用另一高效銅絡合劑,連四硫代鉬酸胺(TTM),可與銅絡合成Cu(MoS4)2自尿液排出,短期內改善癥狀.
3.減少銅吸收 口服鋅制劑可促進肝和腸粘膜細胞合成分泌金屬硫因,與銅離子結合後減少腸銅離子吸收.常用硫酸鋅(每100rug含元素鋅20rug),每日口服量以相當於50mg鋅為宜,分2~3次,餐間服用.對輕癥或病情改善後可單用鋅劑;對病情較重開始治療時,與青黴胺聯合使用,但兩藥須間隔2~3小時,以免療效降低.
4.其他治療 神經系統癥狀可對癥處理,如用左旋多巴、安坦等.肝、腎、骨關節等病癥根據病情適當治療.對本病所致的急性肝功能衰竭或失代償性肝硬化患兒,經上述各種治療無效時可考慮進行肝移植.
小兒肝豆狀核變性饮食
飲食方面要做到規律、合理,即以高蛋白、高維生素食物為主.選擇營養價值高的植物或動物蛋白,如牛奶、蛋類、魚類、瘦肉、各種豆制品等.各種新鮮蔬菜、瓜果富含維生素,營養價值高.
小兒肝豆狀核變性并发症
脾大,肝硬化,錐體束征,癲癇發作,肥胖,高血壓,腎性糖尿,氨基酸尿,蛋白尿,血尿,可出現Fanconi綜合征,發生急性溶血,出血,骨質疏松,骨(軟骨)變性,關節畸形,可致嚴重肝功能衰竭,在數周內死亡,可並發溶血性貧血和失血性貧血,脾功能亢進性貧血等.
1/2 1 2 下一页 尾页


















