魚鱗病 蛇皮癬
魚鱗病 蛇皮癬百科
魚鱗病(ichthyosis)是一組以皮膚幹燥伴片層魚鱗狀粘著性鱗屑為特征的角化異常性遺傳性皮膚病.臨床類型包括尋常型魚鱗病、性聯隱性魚鱗病、先天性非大皰性魚鱗病樣紅皮病、板層狀魚鱗病、先天性大皰性魚鱗病樣紅皮病等.
魚鱗病 蛇皮癬
魚鱗病 蛇皮癬病因
魚鱗病 蛇皮癬
魚鱗病 蛇皮癬症状
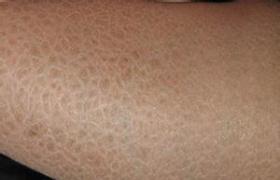
(一)尋常型魚鱗病(ichthyosis vulgaris) 此型最常見,一般冬重夏輕.嬰幼兒即可發病.多累及下肢伸側,尤以小腿最為顯著,四肢屈側及皺褶部位多不累及.病情輕者僅表現為冬季皮膚幹燥,表面有細碎的糠樣鱗屑,又稱幹皮癥(xeroderma);典型皮損是淡褐色至深褐色菱形或多角形鱗屑,鱗屑中央固著,邊緣遊離,如魚鱗狀;臀部及四肢伸側可有毛囊角化性丘疹;掌蹠常見線狀皸裂和掌紋加深;通常無自覺癥狀.
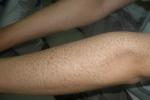
(二)性聯隱性魚鱗病(X-linkedrecessiveichthyosis) 較少見,嬰兒早期發病,僅見於男性,女性僅為攜帶者,一般不發病.可累及全身,以四肢伸側及軀幹下部、脛前明顯,面、頸部亦常受累.表現與尋常型魚鱗病相似,但病情較重,皮膚幹燥粗糙,鱗屑大而顯著,呈黃褐色或污黑色大片魚鱗狀;掌蹠無角化過度.患者可伴隱睪,角膜可有點狀渾濁.病情不隨年齡增長而減輕.
(三)先天性非大皰性魚鱗病樣紅皮病(nonbullouscongenitalichthyosiformerythroderma) 出生時全身皮膚緊張、潮紅,覆有細碎鱗屑,皮膚有緊繃感,面部亦可累及,但眼瞼、口唇外翻少見.隨著年齡增長病情逐漸減輕,至青春期前後趨向好轉.
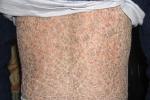
(四)板層狀魚鱗病(lamellarichthyosis) 出生時即出現皮損,表現為黃棕色四方形鱗屑(板層狀),遍及整個體表,嚴重者可似鎧甲樣,以肢體屈側和外陰等處明顯.1/3患者可有眼瞼和口唇外翻,掌蹠常伴角化過度.
(五)先天性大皰性魚鱗病樣紅皮病(bullouscongenitalichthyosiformerythroderma) 出生時即有皮膚潮紅、濕潤和表皮剝脫,受到輕微創傷或摩擦後在紅斑基礎上出現大小不等的薄壁松弛性水皰,易破潰成糜爛面.一般數日後紅斑消退,出現廣泛鱗屑及局限性角化性疣狀丘疹,皮膚皺褶處更明顯,呈“豪豬"樣外觀.新生兒及嬰兒時期常因繼發化膿菌感染而發出臭味,甚至引起敗血癥和水、電解質紊亂而導致死亡.
其他少見的類型還有火棉膠嬰兒與胎兒魚鱗病.

魚鱗病 蛇皮癬
魚鱗病 蛇皮癬检查
魚鱗病 蛇皮癬预防
這個問題確實要引起人們高度重視:該病如在沒有治愈前結婚,對下一代很有可能會遺傳,隻有根治此病才能結婚,平時還要加強鍛煉,對生活方面註意可多飲白開水,服用維生素A.
針對遺傳性魚鱗病,會大大提高子代的遺傳機率,可以結婚,但生育不宜.
魚鱗病 蛇皮癬治疗
魚鱗病 蛇皮癬饮食
(1)忌煙、酒、咖啡、可可.
(2)忌辛辣、燥熱動血的食物.
(3)忌黴變、油煎、肥膩食物.
魚鱗病 蛇皮癬并发症
魚鱗病相關綜合征,大多為常染色體隱性遺傳方式,除累及多個器官和/或組織外,伴有不同程度的魚鱗病樣損害,也是這些綜合征的重要組成部分,臨床癥狀各不相同的IAS,可能是基因多效性的一種表現,魚鱗病的相關綜合征主要有:
①魚鱗病痙攣性癱瘓智能障礙綜合征
為一少見的,由先天性魚鱗病,痙攣性肢體癱瘓和智能障礙三聯癥構成的綜合征,幾乎所有患者出生時就有廣泛的魚鱗病損害,約一半有不同程度的紅皮病,紅皮病隨年齡增長逐步緩解,成年後消失殆盡,終生存在的魚鱗病有3種類型:皺褶部位常為半透明黑色或黑黃色的角化過度損害,皮紋明顯加深;肢體和下腹等處的板層狀魚鱗病損害;其他損害較輕處表現為大量細薄幹燥鱗屑,同一患者常同時具有3種皮損,瞼外翻常見,個別患者毛發幹燥,纖細,無光澤.
大多數患者在出生後4~30個月內發生神經系統病變,主要為智力障礙,痙攣性四肢癱瘓或截癱,雙下肢痙攣性癱瘓進行性加重,至少持續至青春期方緩解或停止進展,多數病人上肢有類似病變,但輕些,絕大多數患者有智能障礙,且不會隨年齡增大而加重.
②魚鱗病侏儒智能障礙綜合征:
這是一個由魚鱗病樣紅皮病,侏儒,智能障礙和痙攣性癱瘓構成的綜合征,在病案記載中:一近親婚配夫妻的5個子女中,發現3例患者,先證者為一18歲女性,身高137cm,出生時面部即有紅皮病表現,雙足背少許大皰,出生數月全身彌漫性發紅,足背等處皮膚萎縮變薄似羊皮紙,16歲後發現雙下肢痙攣性癱瘓,致步行困難,智商38,年齡分別為12歲和8歲的胞妹及胞弟,除無痙攣性癱瘓外,有相似的皮損,智能障礙和侏儒體形.
③智能障礙癲癇魚鱗病綜合征:
嬰兒期發病,主要由輕度的魚鱗病樣紅皮病,智能障礙和癲癇三聯征構成,但有的患者還出現生殖器發育不全,侏儒等癥狀,智能很差,不能自理生活,癲癇多為大發作,本病可能是常染色體隱性遺傳所致,但也不能排除性連鎖遺傳.
④魚鱗病肝脾腫大共濟失調綜合征:
Dykes和Harper(1979~1980)首次報告本病,先證者為一69歲男性,兒童時有全身幹皮病,62歲以後發生進行性加重的構音障礙和步態不穩,幹皮病以肢體伸側最明顯,掌紋很深,肝脾腫大,質硬但肝功能正常,神經系統檢查有老年性癡呆表現,肢體及軀幹運動共濟失調,構音障礙,雙眼向上凝視困難.
⑤毛幹異常智能障礙魚鱗病綜合征:
本病有類似魚鱗病樣紅皮病,發幹異常引起的脆發癥,智能障礙,生育力降低和身材矮小,以發現此類患者中患病傢庭中雙親無同病,男女同患,故認為本病多系常染色體隱性遺傳所致.
⑥魚鱗病多發性神經炎共濟失調綜合征:
為一少見常染色體隱性遺傳性疾病,患者的主要癥狀和體征都緣於神經系統受累,嬰兒期發病,共濟失調表現為步態不穩,意向震顫和眼球震顫,並有四肢軟弱,遠端肌無力,甚至肌萎縮,深發射減弱或消失,此外還可出現神經性耳聾,嗅覺喪失,大多數患者有輕微的魚鱗病皮損,患者多死於急性多發性神經病,腎功能衰竭或心力衰竭.
⑦魚鱗病竹狀發遺傳過敏綜合征:
本病主要由先天性魚鱗病樣紅皮病,竹狀發和遺傳過敏素質三聯構成,罕見,患者多為女性,出生時或出生後不久即有彌漫性紅皮病和脫屑,但是紅皮程度,范圍和持續時間各不相同,魚鱗病臨床表現與局限性線狀魚鱗病無異,頭發稀疏細軟,無光澤,發幹異常,表現為套疊性脆發,狀如竹節故稱竹狀發.
1/2 1 2 下一页 尾页


















